

重测序[1]是指对已知基因组的品种进行个体间的基因组测序,以此来探究不同样本间个体以及群体的差异性。因为其快速简便,检测结果全面且真实可靠,重测序受到了广泛关注。
简单重复序列(simple sequence repeats,SSR)[2,3]又被称作微卫星,是广泛存在于基因组中的1~6个核苷酸串联重复单元,主要包括基因组SSR和表达序列标签SSR。表达序列标签来源于基因的转录区,直接反映基因的表达信息。SSR在真核生物的基因组序列中普遍存在且随机分布。与其他标记相比,SSR标记具有多态性高、重复性好、共显性、易检测、操作简单、无放射、用时短、覆盖面广等优点。利用SSR标记技术建立植物的指纹图谱[4],可以快速且准确鉴定种质资源及品种的纯度。杨育峰等[5]利用甘薯的转录组数据进行SSR标记的开发,对甘薯种质资源进行了多样性分析,构建遗传连锁图谱、分子标记育种和基因挖掘。聂琼等[6]利用烟草同科植物的SSR引物对烟草差异及亲缘关系进行了有效的鉴定分析。陈云等[7]利用SSR分子标记筛选出的6对反应稳定、多态性好的引物将7个烟草品种区分开,并应用于构建马里兰烟的遗传图谱,同时利用其聚类分析结果对烟草类型进行了划分。
基于基因组重测序技术开发遗传分子标记是一种新的方法,尤其对新出现的表现优良的品系进行重测序更加有助于分子育种的发展,有助于优良性状的培育。2014年普通烟草烤烟品种K326、白肋烟品种TN90以及香料烟品种Basma Xanthi的基因组测序工作已经完成[8]。
按烟叶品质特点、生物学性状和栽培调制方法分类,我国将栽培烟草划分为烤烟、晒烟、晾烟、白肋烟、香料烟和黄花烟等6大类。本研究对不同烟草类型共4个烟草品种(系)进行基因组重测序,包括烤烟品系LY1306及烤烟品种秦烟96,晒烟品种为Wanmao 3(WM3H)以及马里兰烟品种为Wufeng 1(WF1H),并结合在NCBI中已发布的烤烟品种K326、白肋烟品种TN90的基因组序列,分析不同烟草类型间以及品种间的InDel突变和SNP突变位点差异,在对数据进行遗传进化分析的基础上筛选准确的遗传标记。
1 材料与方法
1.1 试验样本的采集与DNA测序
试验于2017年在河南农业大学工程楼1413室进行。用于基因组重测序的材料有:烤烟品系LY1306、秦烟96(QY96)和马里兰烟Wufeng 1(WF1H)、晒烟品种Wanmao 3(WM3H),用于试验验证的材料有:烤烟K326与NC89、中烟100、白肋烟TN90与TN86、雪茄烟Cigar 1,均按照常规漂浮育苗法培育幼苗,叶片长至5~6cm时采集烟草叶片样本,液氮速冻,并放-80℃冰箱保存,用于DNA的提取。
参照商业化Qiagen试剂盒DNeasy Plant Mini Kit提取样本的基因组DNA,电泳检测合格(指有清晰条带)后放入-20℃冰箱保存用于建库。电泳检测合格的DNA样本先经过Covaris随机打断成350bp的片段,采用TruSeq DNA LT Sample Prep Kit试剂盒进行建库,DNA片段经过末端修复、加ployA尾、加测序接头、纯化、PCR扩增等步骤,最终完成文库构建。利用PE150测序策略以及Illumina X10平台进行双端测序。为消除测序错误对结果的影响,需要对原始测序数据进行质量控制获取Clean Reads,预处理软件为NGSQC toolkit[9]。数据过滤后将Clean Reads与参考基因组比对,根据比对结果分析SNP和InDel位点差异。
1.2 样本SNP及InDel差异分析
以NCBI网站公布的TN90基因组序列为参考基因组,比较LY1306、QY96、WF1H、 WM3H等4个样本,以及其余2个NCBI上已有参考基因组序列的香料烟品种Basma Xanthi(Basma NCBI)、烤烟品种K326(K326 NCBI)之间的SNP及InDeL差异。
2 结果与分析
2.1 测序数据质量控制及试验样本与参考基因组序列比对
表1 基因组测定序列的质量控制结果
Table 1
| 样本 Sample | 原始数据 Raw Reads data | 有效数据(×108) Clean Reads data | 有效数据比例(%) Clean Reads percent | 原始碱基量 Raw bases data | 有效碱基量 Clean bases data | 有效碱基量比例(%) Clean base data percent | G和C占比(%) G and C percent |
|---|---|---|---|---|---|---|---|
| LY1306 | 974 594 470 | 8.87 | 91.01 | 146189170500 (146.189G) | 132816177340 (132.816G) | 90.85 | 38 |
| QY96 | 985 001 064 | 9.04 | 91.79 | 147750159600 (147.75G) | 135428779630 (135.428G) | 91.66 | 38 |
| WF1H | 993 136 690 | 9.13 | 91.95 | 148970503500 (148.97G) | 136798225077 (136.798G) | 91.82 | 38 |
| M3H | 990 035 276 | 9.17 | 92.67 | 148505291400 (148.505G) | 137424500354 (137.424G) | 92.53 | 38 |
由表1结果可知,4个样本重测序的基因组数据质量控制后的序列有效数据量(Clean Reads data)及有效碱基量(Clean bases data)均达到90%以上,符合测序的质量标准。
表2中质量控制后样品与参考基因组匹配结果表明,各样本参考基因组的匹配率均在99%以上,说明本试验样本经过质量控制后匹配率高,符合质量控制标准。
表2 样本与参考基因组的质量控制匹配结果
Table 2
| 样本 Sample | 有效数据 Clean Reads data | PCR重复比例(%) PCR duplicate rate | 去除PCR重复后数据(×108) Data after PCR duplication were removed | 对比参考基因组的数据(×108) Compare reference genome data | 参考基因组匹配率(%) Reference genome mapping rate |
|---|---|---|---|---|---|
| QY96 | 904 164 146 | 16.18 | 7.58 | 7.56 | 99.74 |
| WM3H | 917 495 832 | 15.53 | 7.75 | 7.74 | 99.91 |
| LY1306 | 887 012 964 | 14.64 | 7.57 | 7.57 | 99.91 |
| WF1H | 913 227 010 | 15.97 | 7.67 | 7.66 | 99.88 |
2.2 试验样本SNP、InDeL检测结果及分析
2.2.1 样本SNP检测结果 以TN90为参照,对比表3中各样本不同位置的突变量,位于基因组区间WF1H突变最少,QY96突变最多,位于非翻译区WF1H突变最少,QY96突变最多,从突变总量来看WF1H最少,QY96最多。通过SNP检测对比发现,样本突变量与其本身有关,QY96不同位点的突变最多,而WF1H不同位点的突变数最少。
表3 各测序品种SNP检测结果
Table 3
| 样本Sample | WF1H | WM3H | LY1306 | QY96 | K326 | BX |
|---|---|---|---|---|---|---|
| 位于基因组上游的突变数The number of mutations in the upstream region | 297 482 | 355 710 | 352 194 | 583 404 | 542 089 | 364 475 |
| 位于基因组下游的突变数The number of mutations in the downstream region | 104 695 | 123 307 | 123 562 | 200 044 | 187 112 | 126 102 |
| 位于基因组区间的突变数The number of mutations in the intergenic region | 3 152 030 | 3 528 347 | 3 463 461 | 5 285 151 | 4 736 983 | 3 386 957 |
| 位于5′端非翻译区域的突变数The number of mutations in the 5′ UTR region | 6 916 | 9 009 | 8 867 | 12 127 | 10 357 | 8 526 |
| 位于3′端非翻译区域的突变数The number of mutations in the 3′ UTR region | 9 323 | 12 474 | 12 396 | 17 041 | 14 327 | 11 539 |
| 位于剪切区域的突变数The number of mutations in the splice region | 4 377 | 5 285 | 5 334 | 6 393 | 5 547 | 5 146 |
| 位于内含子区域的突变数The number of mutations in the intron region | 111 300 | 132 075 | 133 106 | 191 130 | 172 108 | 134 178 |
| 位于外显子区域的突变数The number of mutations in the exon region | 79 199 | 94 325 | 95 240 | 113 782 | 100 511 | 95 749 |
| 总突变数The total mutations | 3 765 322 | 4 260 532 | 4 194 160 | 6 409 072 | 5 769 034 | 4 132 672 |
2.2.2 样本InDeL检测结果 以TN90为参照对比表4中各样本的InDeL突变位点,位于基因组区间WF1H的突变最少,为179 646,K326的突变最多;位于非翻译区的突变WF1H最少,K326最多;从总突变来看,WF1H总突变最小,K326的总突变最多。通过InDeL检测发现,突变数量与样本本身有关,K326不同位点的突变数都为最多,而WF1H不同位点的突变数都是最少。
表4 各测序品种InDeL检测结果
Table 4
| 样本Sample | WF1H | WM3H | LY1306 | QY96 | K326 | BX |
|---|---|---|---|---|---|---|
| 位于基因组上游的突变数The number of mutations in the upstream region | 32 016 | 44 902 | 41 378 | 50 940 | 85 775 | 69 880 |
| 位于基因组下游的突变数The number of mutations in the downstream region | 9 836 | 13 438 | 12 432 | 15 352 | 25 873 | 20 177 |
| 位于基因组区间的突变数The number of mutations in the intergenic region | 179 646 | 226 119 | 212 089 | 250 865 | 437 125 | 295 984 |
| 位于5′端非翻译区域的突变数The number of mutations in the 5′ UTR region | 1 334 | 2 029 | 1 854 | 2 299 | 3 488 | 3 456 |
| 位于3′端非翻译区域的突变数The number of mutations in the 3′ UTR region | 1 104 | 1 604 | 1 405 | 1 998 | 2 968 | 2 241 |
| 位于剪切区域的突变数The number of mutations in the splice region | 662 | 812 | 749 | 874 | 1 081 | 931 |
| 位于内含子区域的突变数The number of mutations in the intron region | 12 614 | 16 880 | 15 448 | 19 796 | 32 146 | 24 199 |
| 位于外显子区域的突变数The number of mutations in the exon region | 2 059 | 2 641 | 2 450 | 3 045 | 4 706 | 3 468 |
| 总突变数The total mutations | 239 271 | 308 425 | 287 805 | 345 169 | 593 162 | 420 336 |
常染色体以50kb划窗,统计窗口内的SNP和InDeL数量,绘制circos图(图1)。统计最长的30条scaffold上的SNP和InDeL数量。
图1
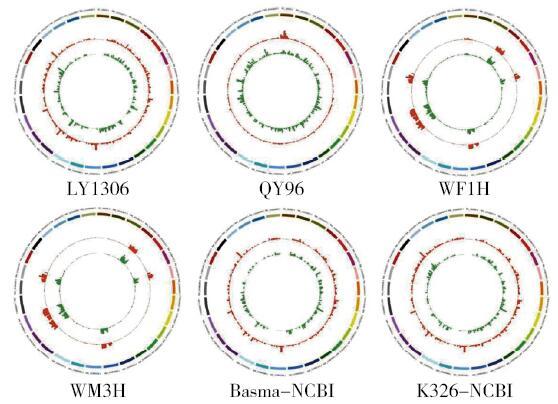
图1
全基因组SNP和InDeL突变位点分布情况
Fig.1
The distribution of mutation sites of SNP and InDeL in the whole genome
通过对不同样本基因组序列差异分析,比较样本间的SNP位点以及InDeL差异基因组序列,4个样本(LY1306、QY96、WM3H、WF1H)的基因组序列差异较大,烤烟与晒烟明显区分,马里兰烟和晒烟亲缘关系较近,同时,LY1306与QY96同为烤烟类型,LY1306与K326亲缘关系较近(图2)。
图2
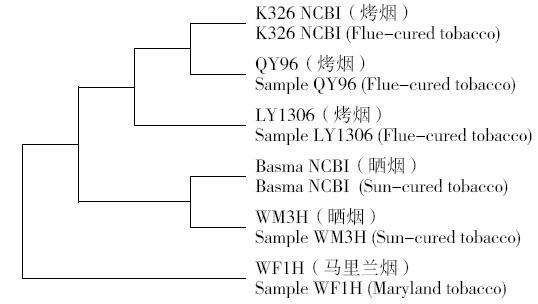
2.3 SSR多态性位点分析
通过对该遗传标记进行试验验证研究,重测序后4个烟草样本(LY1306、QY96、WM3H、WF1H)结合NCBI已有的2个烟草品种(K326、TN90)分析重测序品种(系)的SSR位点,得出7个较为稳定的SSR多态性位点(表5),通过PCR扩增,结果表明,其中5个SSR位点可以扩增出有效片段,是有效的。
表5 基于基因组序列分析SSR多态性位点
Table 5
| 突变位点所在染色体编号 The chromosome number of the mutation site | 突变位点所在染色体上位置 The position of mutation site on the chromosome | 突变位点左端染色体编号 The left end chromosome number of the mutation site | 突变位点右端染色体编号 The right end chromosome number of the mutation site |
|---|---|---|---|
| NW015795276.1 | 14233 | >NW015795276.1:14032-14232 | >NW015795276.1:14233-14433 |
| NW015854676.1 | 34338 | >NW015854676.1:34137-34337 | >NW015854676.1:34338-34538 |
| NW015889872.1 | 10709 | >NW015889872.1:10508-10708 | >NW015889872.1:10709-10909 |
| NW015890969.1 | 7031 | >NW015890969.1:6830-7030 | >NW015890969.1:7031-7231 |
| NW015894675.1 | 7242 | >NW015894675.1:7041-7241 | >NW015894675.1:7242-7442 |
| NW015920095.1 | 7693 | >NW015920095.1:7492-7692 | >NW015920095.1:7693-7893 |
| NW015930318.1 | 14777 | >NW015930318.1:14576-14776 | >NW015930318.1:14777-14977 |
将PCR产物进行琼脂糖凝胶电泳(Marker:DL2000),结果如图3。
图3
表6 用于检测不同烟草类型的SSR多态性位点的引物
Table 6
| 编号No. | 序列名称Seq. name | 正向引物Forward primer (5′→3′) | 反向引物Reverse primer (5′→3′) |
|---|---|---|---|
| 1 | NW015795276.1 | AAAGTGCGTGGAAAAAATC | AGCAGCAGAAGAAAAAGTG |
| 2 | NW015854676.1 | ATCAAATCCATGCTCCCAA | GACCCCAAGATACCCCAAC |
| 3 | NW015889872.1 | CAAAAGCAGCAAAAACACA | GAGAAATGAACGGAAAGTC |
| 4 | NW015890969.1 | CTTAACCACAAAACCCACA | TTCGTAACAATTATCACCG |
| 5 | NW015930318.1 | GTGTTCCTCTGACTTCTCT | CCTTAATCTCAAATCTCTC |
2.4 运用SSR多态性位点检测烟草样本
运用SSR标记对10个烟草样本其中包含了4种烟草类型进行多态性位点检测区分(表7)。
表7 SSR多态性位点对不同烟草类型检测结果
Table 7
| 烟草类型 Tobacco type | 品种(系) Variety (Line) | NW-015795276.1 (205-A 208-B 214-C 220-D) | NW-015854676.1 (114-A 115-B 116-C 120-D 121-E) | NW-015889872.1 (197-A 198-B 199-C 200-D 201-E 202-F) | NW-015890969.1 (168-A 169-B 170-C 171-D 174-E) | NW-015930318.1 (198-A 225-B 231-C 255-D) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 长度Size (bp) | 类型Classification | 长度Size (bp) | 类型Classification | 长度Size (bp) | 类型Classification | 长度Size (bp) | 类型Classification | 长度Size (bp) | 类型Classification | ||||||
| 烤烟 Flue-cured tobacco | K326 | 208 | B | 121 | E | 201 | E | 170 | C | 198 255 | AD | ||||
| NC89 | 205 | A | 121 | E | 201 | E | 170 | C | 198 225 | AB | |||||
| 中烟100 Zhongyan 100 | 205 | A | 116 121 | CE | 200 | D | 170 | C | 198 255 | AD | |||||
| LY1306 | 214 | C | 121 | E | 198 | B | 170 | C | 198 255 | AD | |||||
| QY96 | 205 208 | AB | 121 | E | 201 | E | 170 | C | 231 255 | CD | |||||
| 晾烟(白肋烟) Air-cured tobacco | TN90 | 205 | A | 116 120 | CD | 202 | F | 174 | E | 198 255 | AD | ||||
| (Burley tobacco) | TN86 | 205 | A | 116 120 | CD | 202 | F | 174 | E | 198 225 | AB | ||||
| 晾烟(马里兰烟) Air-cured tobacco (Maryland tobacco) | WF1H | 220 | D | 114 121 | AE | 199 | C | 171 | D | 198 255 | AD | ||||
| 晒烟(雪茄烟) Sun-cured tobacco (Cigar filler) | 茄芯1 Jiaxin1 | 205 | A | 116 120 | CD | 201 | E | 169 | B | 198 255 | AD | ||||
| WM3H | 205 | A | 115 121 | BE | 198 | A | 168 | A | 198 255 | AD | |||||
Note: Size represent the size of the amplified fragment in different species (lines)
注:长度表示在不同品种(系)扩增出来的片段大小
通过对不同类型的烟草重测序后的遗传标记进行试验验证研究,重测序后4个烟草样本(LY1306、QY96、WM3H、WF1H)结合NCBI已有的2个烟草品种(K326、TN90),分析重测序品种的SSR位点以及InDeL位点,得出7个较为稳定的SSR多态性位点,通过PCR片段扩增,结果表明其中5个SSR遗传标记可以有效地把试验品种的遗传多样性和特异性进行区分,将5个SSR标记对10个不同类型烟草样本进行多态性位点的检测区分,结果表明,NW-015795276.1能将马里兰烟与白肋烟、烤烟明显区分,NW-015854676.1能够将烤烟、白肋烟、马里兰烟都分别区分,此外它可以将烤烟品种中的中烟100(ZY100)与其他4个烤烟品种(系)NC89、K326、LY1306、QY96区分开,还可以将晒烟品种Cigar1和WM3H区分开。NW-015889872.1能够将白肋烟和马里兰烟区分开,还可以将烤烟品种(系)中的LY1306和QY96、LY1306和ZY100、ZY100和QY96区分开,可用于区分晒烟中的Cigar1和WM3H。NW-015890969.1也能够将烤烟、白肋烟、马里兰烟明显区分,它还可以区分晒烟中的Cigar1和WM3H。NW-015930318.1能够将NC89、QY96与其余4种烤烟品种(系)区分,还能够将白肋烟品种TN86与TN90区分。NW-015795276.1、NW-015854676.1、NW-015889872.1这3个位点能够将烤烟类型不同品种(系)区分。由以上分析可得,NW-015795276.1、NW-015889872.1和NW-015930318.1可以将烤烟类型LY1306和QY96区分,表明LY1306与QY96虽同为烤烟类型,但其在根本基因序列存在很大差别。NW-015889872.1、NW-015854676.1和NW-015890969.1等3个SSR位点可有效区分烤烟、白肋烟和马里兰烟以及晒烟烟草类型。
3 讨论与结论
SSR标记能准确较快的分析纯度以及对种质资源进行区分,筛选出可用的位点以便分析应用。SSR标记运用于烟草方面起步较晚,且可用的位点较少。陈云等[7]利用聚类分析将马里兰烟和烤烟区分开,证明了SSR标记可用于烟草类型的划分。本试验中QY96和LY1306同为烤烟,同种类型烟草由于外观生长相似,难以区分不同品种(系),SSR遗传标记可以将同类型不同品种(系)烟草(LY306和QY96)进行有效区分。
本研究基于烟草品种(系)重测序,对4个重测序品种(系)进行SNP和InDeL位点分析后得到7个候选SSR位点,并利用PCR技术对包括不同烟草类型在内的多个烟草品种(系)进行了验证,表明其中5个SSR遗传标记可以有效地对不同的烟草类型进行区分。对烤烟品系LY1306和QY96进行遗传鉴定与遗传分析,NW-015795276.1、NW-015889872.1和NW-015930318.1可以有效地将LY1306和QY96区分,NW-015889872.1、NW-015854676.1和NW-015890969.1等3个SSR位点可有效区分烤烟、白肋烟和马里兰烟以及晒烟烟草类型。本研究为烟草新品种(系)的遗传鉴定提供了新的遗传标记位点,在基因遗传水平上研究烤烟品系的差异性,将不同类型的烟草区分开,有效地区分同一类型不同品种(系)间的烟叶样本的差异性和特异性,可以用于对不同品种(系)和类型的烟草进行分类,为烟草遗传鉴定研究提供依据,为不同烟草类型间的鉴别分类提供准确可靠的标记位点,便于进行不同烟草类型间的区分分析。
参考文献
烟草胞质雄性不育系与保持系线粒体DNA的RAPD分析
DOI:10.3969/j.issn.1000-2286.2005.06.006
URL
[本文引用: 1]

用124个10碱基随机引物对烟草2对雄性不育系及其保持系线粒体DNA的RAPD分析表明,不育系与保持系间有112个引物未扩增出多态性片段,占引物总数的92.56%,这反映了不育系和保持系线粒体的同源性。同时,在不育系与保持系的线粒体基因组间也发现了明显的差异,这些差异片段与雄性不育的关系还需进一步研究。
简单重复序列的研究与应用
DOI:10.3969/j.issn.1003-8701.2004.06.003
URL
[本文引用: 1]
SSR是以几个碱基为基本单元的串联重复序列,广泛分布于真核生物基因组中.SSR信息量高,覆盖整个基因组,共显性遗传,多态性水平高,易用PCR分析,是一种非常有用的分子标记.对SSR的种类、研究进展、以及SSR在品种鉴定、基因作图和标记辅助育种等方面的研究应用现状作了简要概述.
简单重复序列的筛选与应用
DOI:10.3969/j.issn.1000-1336.2004.03.028
URL
[本文引用: 1]
综述目前简单重复序列 (SSR) 4种筛选方法的基本原理和主要步骤 ;介绍SSR标记在遗传多样性分析、遗传图谱构建、物种分类和系统发育研究及比较基因组学中的应用
SSR标记及其在蔬菜育种中的应用
DOI:10.3969/j.issn.1009-2196.2005.06.018
URL
[本文引用: 1]
SSR(Simple Sequence Repeats,SSR)是指以1~6个串联核苷酸为单位的重复DNA序列.因每个SSR序列的基本单元重复次数在不同基因型间差异很大,从而形成其座位 的多态性.SSR操作程序包括DNA的提取、DNA的扩增、电泳及染色3个主要环节.SSR技术在蔬菜育种中有着广泛的应用,主要是构建遗传图谱、DNA 指纹图谱的建立、种质资源的遗传多样性等方面.
基于转录组测序数据的甘薯SSR标记开发及群体聚类分析
利用转录组数据开发SSR标记,是目前较为经济高效的DNA分子标记开发方法。为了开发更多适用于甘薯的SSR标记,本研究在前期对甘薯体细胞杂种KT1及其两个亲本4个干旱胁迫处理的24个样本转录组de novo测序的基础上,对获得的105 959条Unigene中大于等于1 000 bp的FASTA序列进行搜索,共找到12 105个SSR位点。从中筛选设计了200对SSR引物在KT1及其亲本中筛选验证,最终选择了20对多态性较好的引物,对来自不同地区的94份甘薯种质资源进行了群体聚类分析。聚类分析把94份实验材料分为3个亚群,较清晰地区分了不同材料之间的遗传相似性。这些基于甘薯转录组测序开发的SSR标记为甘薯的遗传多样性分析、辅助育种和遗传图谱构建等研究的开展提供了更加丰富的候选标记。
23份烟草种质遗传多样性的SSR和ISSR标记分析
DOI:10.3969/j.issn.1001-4829.2011.01.004
URL
[本文引用: 1]
利用SSR和ISSR标记分析 23份烟草种质遗传多样性。结果表明:8个SSR引物和8个ISSR引物分别检测到66个和113个多态性位点。不同烟草种质之间的遗传相异系数在 0.3259~0.8588(SSR)或0.1369~0.8161(ISSR)。以SSR、ISSR标记分别聚类以及两种标记混合聚类均在0.63处将 23个材料分为4类:2个类群和2个独立个类Coker147、Val16或G140,一类群以红花大金元为基础聚类,另一类群以NC82为基础聚类。两 种标记的结果相似,均可用来进行烟草种质资源的遗传多样性分析。
马里兰烟SSR分子标记分析
DOI:10.3969/j.issn.0439-8114.2014.06.057
URL
[本文引用: 2]
采用SSR分子标记分析了五峰7个主栽烟草(Nicotiana tabacum L.)品种.结果表明,6对引物共检测到22个多态位点,7个品种的遗传相似系数为0.347 8~0.9130.
The tobacco genome sequence and its comparison with those of tomato and potato
DOI:10.1038/ncomms4833
URL
PMID:4024737
[本文引用: 1]
The allotetraploid plant Nicotiana tabacum (common tobacco) is a major crop species and a model organism, for which only very fragmented genomic sequences are currently available. Here we report high-quality draft genomes for three main tobacco varieties. These genomes show both the low divergence of tobacco from its ancestors and microsynteny with other Solanaceae species. We identify over 90,000 gene models and determine the ancestral origin of tobacco mosaic virus and potyvirus disease resistance in tobacco. We anticipate that the draft genomes will strengthen the use of N. tabacum as a versatile model organism for functional genomics and biotechnology applications.
NGSQC: cross-platform quality analysis pipeline for deep sequencing data
DOI:10.1186/1471-2164-11-S4-S7
URL
PMID:21143816
[本文引用: 2]
Background While the accuracy and precision of deep sequencing data is significantly better than those obtained by the earlier generation of hybridization-based high throughput technologies, the digital nature of deep sequencing output often leads to unwarranted confidence in their reliability. Results The NGSQC ( N ext G eneration S equencing Q uality C ontrol) pipeline provides a set of novel quality control measures for quickly detecting a wide variety of quality issues in deep sequencing data derived from two dimensional surfaces, regardless of the assay technology used. It also enables researchers to determine whether sequencing data related to their most interesting biological discoveries are caused by sequencing quality issues. Conclusions Next generation sequencing platforms have their own share of quality issues and there can be significant lab-to-lab, batch-to-batch and even within chip/slide variations. NGSQC can help to ensure that biological conclusions, in particular those based on relatively rare sequence alterations, are not caused by low quality sequencing.
Fast and accurate short read alignment with Burrows-Wheeler transform
The sequence alignment/map (SAM) format and SAMtools
DOI:10.1046/j.1440-1665.1999.0178e.x
URL
[本文引用: 1]
PubMed comprises more than 23 million citations for biomedical literature from MEDLINE, life science journals, and online books. Citations may include links to full-text content from PubMed Central and publisher web sites.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}