


小麦是我国三大粮食作物之一,不断提高小麦育种水平对于促进小麦生产发展、保证国家粮食安全具有重要意义。近年来,由于商业化育种对新品种的迫切需要,在小麦杂交育种中主要利用大面积高产品种作为杂交亲本,导致新育成的品种(系)间遗传相似性不断提高,突破性品种越来越少[1]。对小麦品种(系)进行亲缘关系和遗传差异解析,能够更好地了解不同品种(系)的遗传特点,发掘优异的种质材料,对于指导小麦杂交育种的亲本选配、提高小麦育种水平具有重要意义[2,3]。小麦基因组在DNA水平上的研究成果为分析小麦品种间的亲缘关系提供了新的方法和手段。陆续有RAPD[4]、AFLP[5]和SSR[6,7,8,9]等标记应用于小麦种质遗传分析。随着高通量测序技术的出现和发展及后来出现的自动化SNP芯片分型技术,使通过大规模高通量SNP基因芯片进行小麦遗传组成研究越来越普遍。SNP在定位目标基因、构建高密度遗传图谱和群体结构分类等方面具有越来越大的使用价值和发展空间,如小麦遗传图谱的构建[10,11,12,13,14,15]、分子标记辅助选择育种[16,17,18]、全基因组关联分析[19,20,21,22]、遗传多样性[23,24,25]和物种进化[26]研究等方面。曹廷杰等[23]利用Illumina 90k iSelect SNP标记技术分析了河南省自2000年以来近14年审定小麦品种的遗传多样性和遗传基础,结果表明96个小麦品种中,多数品种亲缘关系较近,94.3%的品种间遗传相似系数在0.65~0.81之间;刘易科等[25]利用90K SNP芯片技术对我国主要小麦种植区的240个小麦品种(系)进行全基因组扫描,通过分析参试品种(系)间平均遗传相似系数发现,参试品种(系)间平均遗传相似系数变幅为0.13~0.99,有87.05%的品种(系)遗传相似系数在0.60~0.78之间;聚类分析结果发现我国各省市部分单位因品种(系)交流频繁,育成的品种(系)间出现遗传相似性较高现象。自上世纪90年代至今,山东省泰安市农业科学院在经历了4次育种目标更换,先后育成并审定15个小麦品种,目前还有多个小麦新品系正在参加国家和山东省小麦区域试验,其中以泰山27、泰山28和泰科麦33为代表的小麦品种,在小麦生产中发挥了重要作用。这些小麦品种(系)的遗传基础及遗传多样性差异有待分析明确。利用小麦15 K育种芯片对21个供试小麦品种(系)进行全基因组扫描,分析不同年份育成小麦品种(系)的遗传距离,明确其位点/染色体区段构成差异,揭示全基因组水平上的遗传多样性特点,解析遗传差异的分子基础,以期为小麦新品种组配和选育提供参考依据。
1 材料与方法
1.1 试验材料
供试小麦材料为山东省泰安市农业科学院育成的21个小麦品种(系)。为了探讨不同年份育成小麦品种(系)的遗传构成特点,将21份材料依据育成(审定)年份,分成1990-1999年(90s)期间、2000-2009年(00s)期间、2010-2019年(10s)期间和现在(Present)4个时期进行分析(表1)。
表1 供试小麦材料名称、组合及审(认)定时间
Table 1
| 编号 Number | 品种(系) Variety (line) | 组合 Combination | 审(认) 定年份 Released year | 年代 Age |
|---|---|---|---|---|
| 1 | 鲁麦18 | 86026/8-038//沛县304-1 | 1993 | 90s |
| 2 | 泰山21 | 26744/泰山10号// 鲁麦7号/3/鲁麦18 | 2002/2003 | 00s |
| 3 | 泰山22 | 鲁麦18/鲁麦14 | 2004 | 00s |
| 4 | 泰山23 | 881414/876161 | 2004 | 00s |
| 5 | 泰山24 | 904017/郑州8329 | 2005 | 00s |
| 6 | 泰山9818 | 21-11/935021 | 2006 | 00s |
| 7 | 泰山27 | 泰山651/藏选1号 | 2012 | 10s |
| 8 | 泰山28 | 3262/皖麦38 | 2013 | 10s |
| 9 | 泰科麦30 | 淮阴9908/漯麦9424 | 2019 | 10s |
| 10 | 泰科麦31 | 泰山26/淮麦20 | 2018 | 10s |
| 11 | 泰科麦32 | 洛旱3号/莱州3279 | 2018 | 10s |
| 12 | 泰科麦33 | 郑麦366/淮阴9908 | 2018 | 10s |
| 13 | 泰科紫麦1号 | 良星66/山农紫麦 | 2019 | 10s |
| 14 | 泰科麦38 | 山农17/良星99 | – | Present |
| 15 | 泰科麦308 | 邢麦6号/淮0458 | – | Present |
| 16 | 泰科麦34 | 泰山28/济麦22 | 2020 | Present |
| 17 | 泰科麦6007 | 泰山21/济麦22 | – | Present |
| 18 | 泰科麦36 | 泰农18/齐丰2号 | 2021 | Present |
| 19 | TKM6215 | 良星99/邯94-5378 | – | Present |
| 20 | TKM0564 | 烟农999/良星66 | – | Present |
| 21 | TKM7105 | 泰山26/淮麦0208 | – | Present |
1.2 试验方法
1.2.1 基因组DNA提取 采集供试材料的新鲜叶片组织,利用CTAB法提取基因组DNA。利用琼脂糖凝胶电泳和核酸蛋白测定仪(Nano Drop ND-2000,美国Thermo Fisher Scientific公司)检测DNA质量及浓度,确保达到后续芯片试验要求。
1.2.2 SNP位点分型和质检分析 利用小麦15 K育种芯片对供试小麦品种(系)进行全基因组扫描,在中玉金标记(北京)生物技术股份有限公司平台进行样本DNA的SNP分型,共获得13 947个SNP位点分型数据。为进一步获取高质量的基因分型结果,对数据进行筛选和过滤,选取在芯片检测中Call rate均大于97%的位点标记,过滤后得到10 101个SNP位点的分型结果,剔除基因型检出率小于80%的SNP位点,剩余8692个SNP位点。此外,由于杂合位点在后代会产生分离而干扰检测结果的精密度,因此进一步筛选去除样品中出现杂合分型的位点,最终在所有检测品种(系)中共获得2082个纯合的SNP位点。结合SNP位点在小麦基因组对应位置分析,最终获得2029个定位于小麦21条染色体上的SNP位点。
1.2.3 数据统计分析 结合小麦15 K育种芯片基因型数据信息,将获得的2029个SNP数据分别转换为0(最小等位基因型)和1(相对等位基因型)格式,建立原始矩阵。统计所有SNP位点多态性信息指数(PIC),同时利用SNP分子数据建立的0~1数据矩阵进行每个SNP位点的等位变异丰富度(Aij)和PIC的估算,同时估算A、B和D基因组及全基因组的遗传丰富度(Ri)和多样性指数(Ht),具体计算参考郝晨阳等[27]方法。利用NTSYS-PC ver. 2.2软件中的“DICE”计算品种间遗传相似系数,用非加权配对算术平均法(unweighted pair group method arithmetic averages,UPGMA)绘制遗传关系树状图[28]。用“1-DICE”表示遗传距离,计算不同年代育成品种(系)的平均遗传距离。
1.2.4 基因型图谱构建 结合小麦15 K育种芯片物理位置信息,随机选择均匀分布于全基因组的403个SNP标记(每条染色体约20个)绘制21个小麦品种(系)的基因型图谱。根据以上各SNP位点的等位变异的碱基序列在染色体上标记不同颜色,其中AA标记为红色、TT标记为绿色、CC标记为黄色、GG标记为蓝色、NN(缺失)标记为黑色。利用MapChart 2.32绘制21个小麦品种(系)基因型图谱。
2 结果与分析
2.1 SNP位点的遗传多样性
根据基因分型结果,比较2029个多态性SNP位点在A、B、D 3个基因组、7个同源群和21条染色体的分布特点。结果(表2)表明,2029个SNP基因位点在A、B、D基因组中的分布分别为730、820和479个,A、B、D基因组平均等位变异丰富度分别为2.07、2.14和2.13,平均遗传多样性指数分别为0.24、0.24和0.21。综合2个多样性指标可以看出,B基因组的遗传多样性最高,其次是A和D基因组。由图1可知,在7个同源群中,平均等位变异丰富度依次为H3>H6>H7>H5>H2>H4>H1,平均遗传多样性指数依次为H6>H3>H2>H7>H5>H1>H4。综合2个多样性指数可以看出,二者的分布变化趋势基本一致,第3和第6同源群具有较高的遗传多样性,第1和第4同源群的遗传多样性较低。
表2 3个基因组的遗传多样性比较
Table 2
| 项目Item | A基因组 A genome | B基因组 B genome | D基因组 D genome | 合计 Total | 均值 Mean |
|---|---|---|---|---|---|
| 检测位点数Number of the detected locus | 730 | 820 | 479 | 2029 | – |
| 等位变异总数(∑Aij)Total allelic variations | 1510 | 1753 | 1022 | 4285 | – |
| 平均等位变异丰富度(∑Aij/Loci)Mean allele richness | 2.07±0.0006 | 2.14±0.0005 | 2.13±0.0011 | – | 2.11±0.0010 |
| 平均遗传多样性指数(Ht)Mean genetic diversity index | 0.24±0.0002 | 0.24±0.0002 | 0.21±0.0003 | – | 0.23±0.0002 |
遗传多样性各指标的标准差计算参考文献[
The standard deviation of genetic diversity is calculated according to the reference [29], the same below
图1
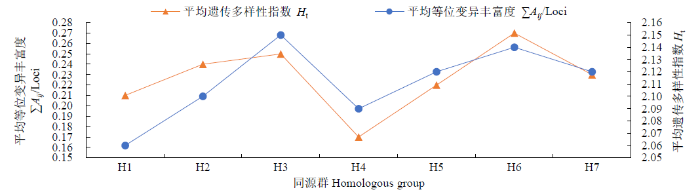
图1
育成品种(系)7个同源群的遗传多样性
Fig.1
Genetic diversity of seven homologous groups of the varieties (lines)
由图2可知,在21条染色体中,A基因组的1A染色体的变异丰富度和遗传多样性指数均很低,6A的平均等位变异丰富度偏低;B基因组染色体的平均等位变异丰富度和多样性指数均较高,且2个指标变化趋势基本一致;D基因组的2D和6D染色体的平均等位变异丰富度较高。整体来看,3A、1B、6B染色体的遗传多样性较高,而1A、6A的遗传多样性偏低。
图2
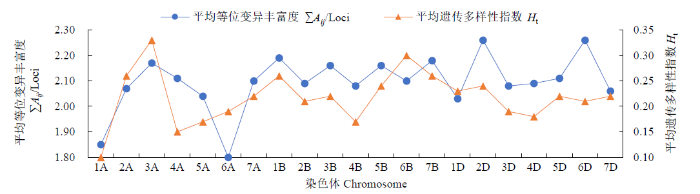
图2
育成品种(系)21条染色体的遗传多样性
Fig.2
Genetic diversity of 21 chromosomes of varieties (lines)
2.2 供试材料的遗传距离分析
结果(图3)表明,不同年份小麦品种(系)平均遗传距离的变异范围为0.00~0.37。从平均遗传距离的变化曲线来看,自90s以来,遗传距离总体呈先升后降趋势。由于只有1个90s育成品种,其平均遗传距离为0.00,00s品种(系)间的平均遗传距离为0.10,10s品种(系)间平均遗传距离出现最高值0.37后,现在(Present)品种(系)的平均遗传距离为0.07,降到最低。
图3
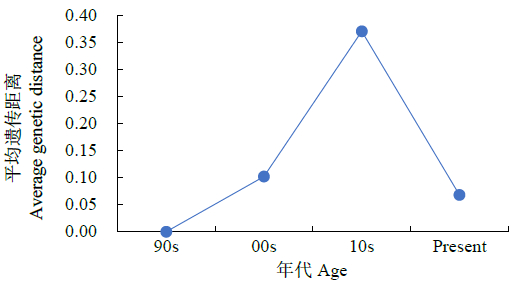
图3
不同年代选育品种(系)的平均遗传距离
Fig.3
Average genetic distance of wheat varieties (lines) in different ages
2.3 供试材料的聚类分析
为明确21个小麦品种(系)之间的亲缘关系及与系谱关系是否吻合,对其进行聚类分析,结果(图4)表明,21个供试小麦品种(系)间的遗传相似系数在0.69~0.99之间。除早熟高产品种泰山24、优质品种泰山27、高产品种泰科麦30、高产节水型品种泰科麦32和高产优质品种泰科麦33外,其余品种(系)可以分为4个类群。90s育成的早熟高产品种鲁麦18和00s育成的早熟超高产品种泰山23归为类群Ⅰ;00s育成的早熟高产品种泰山21、大穗高产品种泰山9818和早熟高产优质品种泰山22归为类群Ⅱ;类群Ⅲ为10s和现在育成的小麦品种(系),包括高产品种泰科麦31和特用品种泰科紫麦1号,高产节水品系泰科麦38、高产品系泰科麦308、早熟品系泰科麦6007、优质品系泰科麦36、高产品系TKM0564和TKM7105等;类群Ⅳ主要为10s和现在育成品种(系),包括高产品种泰山28、高产新品系泰科麦34和优质高产抗倒新品系TKM6215,基本上同一年份育成的品种(系)聚在一起,跟系谱关系(表1)较为吻合。
图4
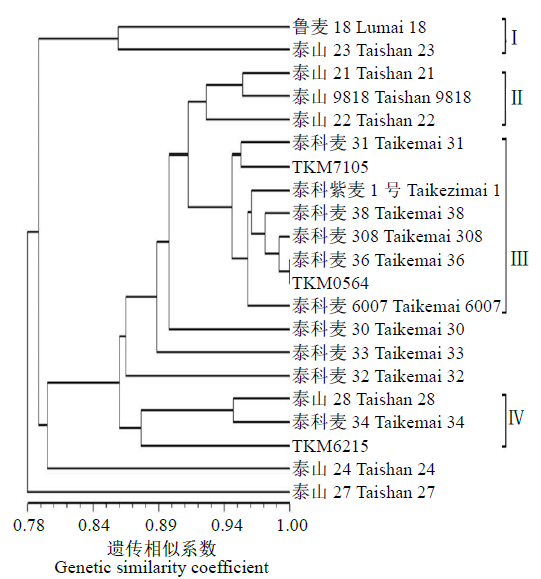
2.4 供试材料的共有位点/染色体区段
根据2029个定位的纯合SNP标记的物理位置随机选取均匀分布染色体的403个SNP标记(每条染色体约20个),绘制21个供试小麦品种(系)的基因型图谱。
表3和图5表明,在不同的年份(00s、10s和Present)育成小麦品种(系)间的共有SNP的数量、基因组分布和同源群分布均有差异。00s育成的5个小麦品种携带的共有SNP为203个,主要分布在A、B基因组和H4、H7同源群;10s育成的7个小麦品种携带的共有SNP为122个,主要分布在D和A基因组和H2、H6同源群;Present育成的8个小麦品种(系)携带的共有SNP为251个,主要分布在A和B基因组和H2、H3同源群。分析同一年份育成的品种(系)共有的染色体区段情况发现:00s育成的5个小麦品种携带的相同染色体区段主要分布在1A、2A、7A、3B、4B、6B和4D染色体上,共包含57个位点;10s育成的7个小麦品种携带的相同染色体区段主要分布在1A、2A、2D、5D、6B、6D和7D染色体上,共包含53个位点;Present育成的8个小麦品种(系)携带的相同染色体区段主要分布在2A、4A、7A、2B、3B、4B、6B和6D染色体上,共包含109个位点(附表1)。
表3 不同育种年份间共有的SNP数量、基因组(A、B、D)分布和同源群(H1~H7)分布
Table 3
| 名称 Name | 年代Age | ||
|---|---|---|---|
| 00s | 10s | Present | |
| SNP | 203 | 122 | 251 |
| A | 74 | 34 | 87 |
| B | 72 | 33 | 88 |
| D | 57 | 55 | 76 |
| H1 | 19 | 14 | 28 |
| H2 | 27 | 31 | 46 |
| H3 | 32 | 10 | 42 |
| H4 | 37 | 9 | 38 |
| H5 | 29 | 15 | 30 |
| H6 | 24 | 30 | 34 |
| H7 | 35 | 13 | 33 |
图5

图5
21个小麦品种(系)的基因型图谱
箭头标记的是21个小麦品种(系)共有SNP位点的位置
Fig.5
Genotypic maps of 21 wheat varieties (lines)
The arrows mark the SNP loci owned by all of 21 wheat varieties (lines)
进一步比较21个供试小麦品种(系)基因型图谱,发现含有25个共同的SNP位点(图5),这些共有SNP位点分布在1A、5A、6A、7A、2B、3B、6B、1D、2D、3D和7D染色体上,且每条染色体上分布SNP位点的数目均不相同,其中1A染色体上的AX-111786969-AX、109920244-AX、110049336、AX-110094282和AX-109335959位点成簇分布。
3 讨论
本研究结果表明不同年份小麦品种(系)平均遗传距离呈先升高后下降的趋势。在90s,因仅育成1个小麦品种,平均遗传距离为0.00,无法比较;00s育成品种的平均遗传距离为0.10,可能与较多地利用鲁麦14[32]、鲁麦18及其衍生后代作为杂交亲本有关;10s育成品种平均遗传距离为0.37,达到最高值,在亲本的利用上引入了亲缘关系相对较远的优质材料;现在育成品种(系)间的平均遗传距离出现下降趋势,可能与较多地利用大面积高产品种作为杂交亲本有一定关系。本研究的聚类分析结果可将供试材料大致分为4个类群,且同一年份的品种(系)基本聚在一起,与系谱分析结果吻合。例如,类群Ⅰ的2个品种亲本86026和881414亲缘关系较近;类群Ⅱ中的泰山21和泰山22的亲本之一都是鲁麦18;类群Ⅲ的8个品种(系)中,泰科麦31和TKM7105的共同亲本为泰山26,泰科麦0311和TKM0564的亲本均含有烟麦系列的血缘,泰科紫麦1号和泰科麦38均含有良星系列的亲本。另外,未被聚类的5个育成小麦品种(系)的亲本中含有郑州8329、藏选1号、漯麦9424、洛旱3号、莱州3279、郑麦366和淮阴9908等的遗传区段或位点,与前面4个类群所用亲本的亲缘关系较远。
目前育成小麦品种(系)间的遗传相似性较高,遗传多样性降低已成为一种普遍现象。郝晨阳等[27]通过分析我国育成品种的遗传多样性,发现从20世纪50年代起,我国小麦育成品种的遗传多样性指数和品种间平均遗传距离逐步下降;王江春等[6]研究表明,山东省小麦品种遗传距离总体呈下降趋势,50年代品种间的遗传距离最大,之后逐渐降低,80年代达到最低,90年代出现一个高值后又明显降低;程斌等[33]选用53个SSR引物对96个小麦品种(系)进行了遗传多样性分析,证明山东省近年来育成小麦品种(系)遗传基础越来越狭窄、多态性呈下降的趋势。因此,拓宽小麦品种的遗传基础,提高育成品种的遗传多样性,是今后小麦品种遗传改良研究中值得重视和亟待解决的问题。
通过分析不同年份育成小麦品种(系)共有SNP和共有染色体区段基因组分布情况发现,00s、10s和Present育成的小麦品种(系)共有SNP和共有染色体区段分别主要分布在A、D和B基因组。查阅相关文献发现A基因组含有丰富的与磷高效[34]、株高[35,36]、千粒重[37]、抽穗期[38]、分蘖数[39]、Glu-A3[40]、产量[41]和籽粒性状[42]等重要性状相关的基因;B基因组含有丰富的与株高[36,43]、穗长[44]、分蘖数[34]、抽穗期[45,46,47]、成熟期[37,45]、磷高效[34]、籽粒/面粉蛋白质[48]和抗逆[15,41,49]等重要性状相关的基因;D基因组含有丰富的植物抗逆[15,38,49]、高产[41,50]和优质[51,52]等优异基因。以上结果与本文不同年份育种目标吻合,如00s的高产早熟目标、10s高产抗逆目标及现在的高产优质、多抗和节本安全等目标。同时本研究发现21份供试材料均含有25个共同的SNP位点,分布在1A、5A、6A、7A、2B、3B、6B、1D、2D、3D和7D染色体上。通过对比已报道的小麦产量等QTL信息[15, 34, 36-37, 41-43, 45-46, 49-50, 53-58],并选取物理位置10Mb之内的QTL分析发现,25个SNP位点与增加产量的QTL(如产量、千粒重、穗粒数和单株穗数等)、控制株高的QTL、增加分蘖数的QTL、控制开花期和抽穗期的QTL、提高叶锈病和白粉病抗性的QTL以及提高灌浆速率的QTL等存在密切关系。以上供试材料的遗传分析结果可能与品种选育过程中注重选择携带控制重要性状的关键基因有关,所以在杂种后代中受到较多的选择和保留。
4 结论
21个小麦品种(系)间的遗传差异日趋缩小,遗传多样性逐渐降低;同一年份的品种(系)基本聚在同一类群,与其系谱关系吻合;同一年份育成的小麦品种(系)携带相同的SNP或染色体区段与不同年份的育种目标吻合;在品种(系)组配与选育过程中注重产量、株高、分蘖数、抽穗期、灌浆速率和抗病等性状的选择,可为今后小麦新品种选育提供参考依据。
参考文献
Genetic relationships and diversity among Tibetan wheat,common wheat and European spelt wheat revealed by RAPD markers
DOI:10.1023/A:1018316129246 URL [本文引用: 1]
Quantitative evaluation of genetic diversity in Iranian modern cultivars of wheat (Triticum aestivum L.) using morphological and amplified fragment length polymorphism (AFLP) markers
Assessment of genetic diversity of Yunnan,Tibetan,and Xinjiang wheat using SSR markers
DOI:10.1016/S1673-8527(07)60071-X URL [本文引用: 1]
Development of a high- density SNP- based linkage map and detection of yellow pigment content QTLs in durum wheat
DOI:10.1007/s11032-014-0183-3 URL [本文引用: 1]
Characterization of a wheat breeder’s array suitable for high-throughput SNP genotyping of global accessions of hexaploid bread wheat (Triticum aestivum)
DOI:10.1111/pbi.2017.15.issue-3 URL [本文引用: 1]
Ultra-dense genetic map of durum wheat × wild emmer wheat developed using the 90K iSelect SNP genotyping assay
DOI:10.1007/s11032-014-0176-2 URL [本文引用: 1]
High-throughput SNP genotyping of modern and wild emmer wheat for yield and root morphology using a combined association and linkage analysis
Mapping of QTL for partial resistance to powdery mildew in two Chinese common wheat cultivars
DOI:10.1007/s10681-019-2537-8 URL [本文引用: 5]
A SNP marker for the selection of HfrDrd,a Hessian fly- response gene in wheat
DOI:10.1007/s11032-015-0410-6 URL [本文引用: 1]
Genotyping-by-sequencing (GBS)identified SNP tightly linked to QTL for pre-harvest sprouting resistance
DOI:10.1007/s00122-015-2513-1 URL [本文引用: 1]
Development of genotyping by sequencing (GBS)- and array-derived SNP markers for stem rust resistance gene Sr42
DOI:10.1007/s11032-015-0404-4 URL [本文引用: 1]
Genome-wide association mapping for seedling and field resistance to Puccinia striiformis f.sp. tritici in elite durum wheat
DOI:10.1007/s00122-016-2841-9 URL [本文引用: 1]
Genome-wide DArT and SNP scan for QTL associated with resistance to stripe rust (Puccinia striiformis f. sp. tritici) in elite ICARDA wheat (Triticum aestivum L.) germplasm
DOI:10.1007/s00122-015-2504-2 URL [本文引用: 1]
Genome-wide association study for kernel weight-related traits using SNPs in a Chinese winter wheat population
DOI:10.1007/s10681-016-1750-y URL [本文引用: 1]
Genetic diversity and phenetic analysis in wheat (Triticum turgidum subsp. durum and Triticum aestivum subsp. aestivum) landraces based on SNP markers
DOI:10.1007/s10722-016-0435-7 URL [本文引用: 1]
Population- and genome-specific patterns of linkage disequilibrium and SNP variation in spring and winter wheat (Triticum aestivum L.)
DOI:10.1186/1471-2164-11-727 URL [本文引用: 2]
NTSYS-pc:Numerical taxonomy and multivariate analysis system. Version 2.2
Sampling variance of the genetic diversity index
DOI:10.1093/oxfordjournals.jhered.a110169 URL [本文引用: 1]
SNP-revealed genetic diversity in wild emmer wheat correlates with ecological factors
DOI:10.1186/1471-2148-13-169 URL [本文引用: 1]
Population structure,genetic diversity and linkage disequilibrium in elite winter wheat assessed with SNP and SSR markers
DOI:10.1007/s00122-013-2065-1
PMID:23429904
[本文引用: 1]

Modern genomics approaches rely on the availability of high-throughput and high-density genotyping platforms. A major breakthrough in wheat genotyping was the development of an SNP array. In this study, we used a diverse panel of 172 elite European winter wheat lines to evaluate the utility of the SNP array for genomic analyses in wheat germplasm derived from breeding programs. We investigated population structure and genetic relatedness and found that the results obtained with SNP and SSR markers differ. This suggests that additional research is required to determine the optimum approach for the investigation of population structure and kinship. Our analysis of linkage disequilibrium (LD) showed that LD decays within approximately 5-10 cM. Moreover, we found that LD is variable along chromosomes. Our results suggest that the number of SNPs needs to be increased further to obtain a higher coverage of the chromosomes. Taken together, SNPs can be a valuable tool for genomics approaches and for a knowledge-based improvement of wheat.
Detection of QTLs for phosphorus use efficiency in relation to agronomic performance of wheat grown under phosphorus sufficient and limited conditions
DOI:10.1016/j.plantsci.2009.03.006 URL [本文引用: 4]
Genetic dissection of yield-related traits in a recombinant inbred line population created using a key breeding parent in China’s wheat breeding
DOI:10.1007/s00122-013-2123-8 URL [本文引用: 3]
QTL mapping for grain filling rate and yield-related traits in RILs of the Chinese winter wheat population Heshangmai × Yu8679
DOI:10.1007/s00122-008-0901-5
PMID:18853131
[本文引用: 3]
A set of 142 winter wheat recombinant inbred lines (RILs) deriving from the cross Heshangmai x Yu8679 were tried in four ecological environments during the seasons 2006 and 2007. Nine agronomic traits comprising mean grain filling rate (GFR(mean)), maximum grain filling rate (GFR(max)), grain filling duration (GFD), grain number per ear (GNE), grain weight per ear (GWE), flowering time (FT), maturation time (MT), plant height (PHT) and thousand grain weight (TGW) were evaluated in Beijing (2006 and 2007), Chengdu (2007) and Hefei (2007). A genetic map comprising 173 SSR markers and two EST markers was generated. Based on the genetic map and phenotypic data, quantitative trait loci (QTL) were mapped for these agronomic traits. A total of 99 putative QTLs were identified for the nine traits over four environments except GFD, PHT and MT, measured in two environments (BJ07 and CD07), respectively. Of the QTL detected, 17 for GFR(mean), 16 for GFR(max), 21 for TGW and 10 for GWE involving the chromosomes 1A, 1B, 2A, 2D, 3A, 3B, 3D, 4A, 4D, 5A, 5B, 6D and 7D were identified. Moreover, 13 genomic regions showing pleiotropic effects were detected in chromosomes 1A, 1B, 1D, 2A, 2B, 2D, 3A, 3B, 4B, 4D, 5B, 6D and 7D; these QTL revealing pleiotropic effects may be informative for a better understanding of the genetic basis of grain filling rate and other yield-related traits, and represent potential targets for multi-trait marker aided selection in wheat.
Advanced backcross QTL analysis of a hard winter wheat × synthetic wheat population
Advanced backcross quantitative trait locus (AB-QTL) analysis was used to identify QTLs for yield and yield components in a backcross population developed from a cross between hard red winter wheat (Triticum aestivum L.) variety Karl 92 and the synthetic wheat line TA 4152-4. Phenotypic data were collected for agronomic traits including heading date, plant height, kernels per spike, kernel weight, tiller number, biomass, harvest index, test weight, grain yield, protein content, and kernel hardness on 190 BC2F(2:4) lines grown in three replications in two Kansas environments. Severity of wheat soil-borne mosaic virus (WSBMV) reaction was evaluated at one location. The population was genotyped using 151 microsatellite markers. Of the ten putative QTLs identified, seven were located on homologous group 2 and group 3 chromosomes. The favorable allele was contributed by cultivated parent Karl 92 at seven QTLs including a major one for WSBMV resistance, and by the synthetic parent at three QTLs: for grain hardness, kernels per spike, and tiller number.
QTL mapping for yield and yield contributing traits in two mapping populations of bread wheat
DOI:10.1007/s11032-006-9056-8 URL [本文引用: 1]
Characterization and marker development for low molecular weight glutenin genes from Glu-A3 alleles of bread wheat (Triticum aestivum L.)
PCR was used to amplify low-molecular-weight (LMW) glutenin genes from the Glu-A3 loci of hexaploid wheat cultivars containing different Glu-A3 alleles. The complete coding sequence of one LMW glutenin gene was obtained for each of the seven alleles Glu-A3a to Glu-A3g. Chromosome assignment of PCR products using Chinese Spring nulli-tetrasomic lines confirmed the amplified products were from chromosome 1A. All sequences were classified as LMW-i-type genes based on the presence of an N-terminal isoleucine residue and eight cysteine residues located within the C-terminal domain of the predicted, mature amino acid sequence. All genes contained a single uninterrupted open reading frame, including the sequence from the Glu-A3e allele, for which no protein product has been identified. Comparison of LMW glutenin gene sequences obtained from different alleles showed a wide range of sequence identity between the genes, with between 1 and 37 single nucleotide polymorphisms and between one and five insertion/deletion events between genes from different alleles. Allele-specific PCR markers were designed based on the DNA polymorphisms identified between the LMW glutenin genes, and these markers were validated against a panel of cultivars containing different Glu-A3 alleles. This collection of markers represents a valuable resource for use in marker-assisted breeding to select for specific alleles of this important quality-determining locus in bread wheat.
Genetics of yield,abiotic stress tolerance and biofortification in wheat (Triticum aestivum L.)
DOI:10.1007/s00122-020-03583-3 URL [本文引用: 4]
QTL analysis of wheat kernel traits,and genetic effects of qKW-6A on kernel width
DOI:10.1007/s10681-018-2333-x URL [本文引用: 1]
Genomic regions for yield and yield parameters in Chinese winter wheat (Triticum aestivum L.) genotypes tested under varying environments correspond to QTL in widely different wheat materials
DOI:10.1016/j.plantsci.2008.03.006 URL [本文引用: 2]
QTL analysis of spike morphological traits and plant height in winter wheat (Triticum aestivum L.) using a high-density SNP and SSR-based linkage map
Molecular mapping of quantitative trait loci for yield and yield components in spring wheat (Triticum aestivum L.)
DOI:10.1007/s00122-008-0804-5
PMID:18516583
[本文引用: 3]
An F1 derived doubled haploid (DH) population of 402 lines from the adapted spring wheat cross Superb (high yielding)/BW278 (low yielding) was developed to identify quantitative trait loci (QTL) associated with yield and yield components. A subset of the population (186 lines) was evaluated in replicated field trials in 2001 and 2002 at six locations in Manitoba and Saskatchewan, Canada. Agronomic parameters, grain yield and yield components including 1,000 grain weight, harvest index, average seed weight spike(-1), seed number spike(-1) and spikes number m(-2) were measured. A genetic map was constructed with 268 microsatellite marker loci and included two morphological genes, reduced plant height, Rht-B1b, and the presence/absence of awns, B1. Composite interval mapping was conducted to estimate the location and effect of QTL associated with the evaluated traits. A total of 53 QTL were identified on 12 chromosomes for the 9 evaluated traits with the coefficient of determination ranging from 0.03 to 0.21 of the total variation. The increase in yield and yield components ranged from 4.5 to 17.1% over the population mean. The five grain yield QTL were detected on chromosomes 1A, 2D, 3B, and 5A and showed a combined increase of 34.4%, over the population mean. The alleles from Superb were associated with increased yield for four of the five QTL. This study identified potential chromosome segments for use in marker-assisted selection to improve yield and yield components in spring wheat.
Mapping QTLs with main and epistatic effects underlying grain yield and heading time in soft winter wheat
DOI:10.1007/s00122-011-1583-y URL [本文引用: 2]
Pleiotropic QTL influencing spikelet number and heading date in common wheat (Triticum aestivum L.)
DOI:10.1007/s00122-020-03556-6 URL [本文引用: 1]
Molecular detection of QTLs for agronomic and quality traits in a doubled haploid population derived from two Canadian wheats (Triticum aestivum L.)
Development of high-yielding wheat varieties with good end-use quality has always been a major concern for wheat breeders. To genetically dissect quantitative trait loci (QTLs) for yield-related traits such as grain yield, plant height, maturity, lodging, test weight and thousand-grain weight, and for quality traits such as grain and flour protein content, gluten strength as evaluated by mixograph and SDS sedimentation volume, an F1-derived doubled haploid (DH) population of 185 individuals was developed from a cross between a Canadian wheat variety "AC Karma" and a breeding line 87E03-S2B1. A genetic map was constructed based on 167 marker loci, consisting of 160 microsatellite loci, three HMW glutenin subunit loci: Glu-A1, Glu-B1 and Glu-D1, and four STS-PCR markers. Data for investigated traits were collected from three to four environments in Manitoba, Canada. QTL analyses were performed using composite interval mapping. A total of 50 QTLs were detected, 24 for agronomic traits and 26 for quality-related traits. Many QTLs for correlated traits were mapped in the same genomic regions forming QTL clusters. The largest QTL clusters, consisting of up to nine QTLs, were found on chromosomes 1D and 4D. HMW glutenin subunits at Glu-1 loci had the largest effect on breadmaking quality; however, other genomic regions also contributed genetically to breadmaking quality. QTLs detected in the present study are compared with other QTL analyses in wheat.
QTL mapping of adult-plant resistance to leaf rust in the Chinese landraces Pingyuan 50/Mingxian 169 using the wheat 55K SNP array
DOI:10.1007/s11032-019-1004-5 URL [本文引用: 3]
Genetic mapping of quantitative trait loci associated with Important agronomic traits in the spring wheat (Triticum aestivum L.) cross ‘Louise’ × ‘Penawawa’
DOI:10.2135/cropsci2010.03.0185 URL [本文引用: 2]
Identification of three Wx proteins in wheat (Triticum aestivum L.)
Nullisomic analysis of waxy (Wx) protein of hexaploid wheat (Triticum aestivum L.) cv. "Chinese Spring" using two-dimensional polyacrylamide gel electrophoresis revealed that three Wx loci, Wx-A1, Wx-B1, and Wx-D1, located on chromosome arms 7AS, 4AL, and 7DS, produce three distinct Wx subunit groups, subunit group-A (SGA), SGB, and SGD, respectively. SGA has a higher molecular weight and a more basic isoelectric point (pI) than the other two. SGB and SGD have the same molecular weight but a slightly different pI range. Owing to the detection of these three subunit groups, we were able to identify the expression of three waxy genes in wheat endosperm and to find two types of mutants among Japanese wheat cultivars, one lacking SGA and the others SGB. These results suggest the possibility of breeding a waxy wheat.
Allelic variation of polyphenol oxidase (PPO) genes located on chromosomes 2A and 2D and development of functional markers for the PPO genes in common wheat
Polyphenol oxidase (PPO) activity is highly related to the undesirable browning of wheat-based end products, especially Asian noodles. Characterization of PPO genes and the development of their functional markers are of great importance for marker-assisted selection in wheat breeding. In the present study, complete genomic DNA sequences of two PPO genes, one each located on chromosomes 2A and 2D and their allelic variants were characterized by means of in silico cloning and experimental validation. Sequences were aligned at both DNA and protein levels. Two haplotypes on chromosome 2D showed 95.2% sequence identity at the DNA level, indicating much more sequence diversity than those on chromosome 2A with 99.6% sequence identity. Both of the PPO genes on chromosomes 2A and 2D contain an open reading frame (ORF) of 1,731 bp, encoding a PPO precursor peptide of 577 amino acids with a predicted molecular mass of approximately 64 kD. Two complementary dominant STS markers, PPO16 and PPO29, were developed based on the PPO gene haplotypes located on chromosome 2D; they amplify a 713-bp fragment in cultivars with low PPO activity and a 490-bp fragment in those with high PPO activity, respectively. The two markers were mapped on chromosome 2DL using a doubled haploid population derived from the cross Zhongyou 9507/CA9632, and a set of nullisomic-tetrasomic lines and ditelosomic line 2DS of Chinese Spring. QTL analysis indicated that the PPO gene co-segregated with the two STS markers and was closely linked to SSR marker Xwmc41 on chromosome 2DL, explaining from 9.6 to 24.4% of the phenotypic variance for PPO activity across three environments. In order to simultaneously detect PPO loci on chromosomes 2A and 2D, a multiplexed marker combination PPO33/PPO16 was developed and yielded distinguishable DNA patterns in a number of cultivars. The STS marker PPO33 for the PPO gene on chromosome 2A was developed from the same gene sequences as PPO18 that we reported previously, and can amplify a 481-bp and a 290-bp fragment from cultivars with low and high PPO activity, respectively. A total of 217 Chinese wheat cultivars and advanced lines were used to validate the association between the polymorphic fragments and grain PPO activity. The results showed that the marker combination PPO33/PPO16 is efficient and reliable for evaluating PPO activity and can be used in wheat breeding programs aimed for noodle and other end product quality improvement.
Identifying loci influencing grain number by microsatellite screening in bread wheat (Triticum aestivum L.)
DOI:10.1007/s00425-012-1708-9 URL
Advanced backcross QTL analysis in progenies derived from a cross between a German elite winter wheat variety and a synthetic wheat (Triticum aestivum L.)
PMID:15243706
We report here the second advanced backcross quantitative trait locus (AB-QTL) analysis carried out in winter wheat. Seven agronomic traits were studied in a BC2F1 population derived from a cross between the German winter wheat variety Flair and the synthetic wheat line XX86 developed in Japan. We selected 111 BC2F1 lines and genotyped these with 197 microsatellite markers. Field data for seven agronomic traits were collected from corresponding BC2F3 families that were grown at up to six locations in Germany. QTL analyses for yield and yield components were performed using single-marker regression and interval mapping. A total of 57 putative QTLs derived from XX86 were detected, of which 24 (42.1%) were found to have a positive effect from the synthetic wheat XX86. These favourable QQTLs were mainly associated with thousand-grain weight and grain weight per ear. Many QTLs for correlated traits were mapped in similar chromosomal regions. The AB-QTL data obtained in the present study are discussed and compared with results from previous QTL analyses.
QTL mapping and successful introgression of the spring wheat‑derived QTL Fhb1 for Fusarium head blight resistance in three European triticale populations
DOI:10.1007/s00122-019-03476-0 URL
Mapping quantitative trait loci for quality factors in an inter-class cross of US and Chinese wheat
DOI:10.1007/s00122-009-1232-x URL
QTL analysis of kernel shape and weight using recombinant inbred lines in wheat
DOI:10.1007/s10681-008-9794-2 URL [本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}