

我国普通菜豆主产区主要分布在黑龙江、内蒙古、吉林、云南、四川、贵州、山西、陕西、甘肃等土地相对贫瘠的干旱半干旱地区,因此改良普通菜豆根系对提高水分、养分等的利用率具有重要意义。有关普通菜豆根系性状的研究较多,但主要集中在根系生理特性及养分吸收等方面[12,13,14,15,16],根系性状的遗传研究相对较少。以往的研究在不同逆境下均检测到与根长、根干重、根角度等相关的QTL[9,10,11],但大多是生育后期的相关性状,缺少对幼苗期根系性状的研究。有研究认为,生育早期良好的根系发育可以促进根系对营养元素的吸收[2],对增产有积极作用,幼苗期根系发达、生长速率快是作物抗逆的主要根系特征[17]。因此,对普通菜豆幼苗期根系进行遗传研究,发掘与其紧密连锁的分子标记,对普通菜豆的高产育种和抗逆改良均具有重要意义。本研究以324份普通菜豆种质为材料,结合116对多态性SSR标记,对幼苗期根系性状进行关联分析,发掘与普通菜豆幼苗期根系紧密关联的分子标记,为普通菜豆根系的分子标记辅助育种改良提供理论依据。
1 材料与方法
1.1 试验材料
试验材料由中国农业科学院作物科学研究所提供,来源为我国普通菜豆主产区黑龙江、山西、陕西、内蒙古等省区的地方品种,以及来自美国、哥伦比亚、巴西等国家和国际热带农业研究中心的引进品种(表1)。
表1 供试普通菜豆材料
Table 1
| 统一编号Number | 种质Germplasm | 来源Origin | 统一编号Number | 种质Germplasm | 来源Origin | |
|---|---|---|---|---|---|---|
| F0002119 | 白小豆 | 中国甘肃 | - | 刀豆 | 中国山东 | |
| F0002120 | 白小豆 | 中国甘肃 | F0000045 | 洋菜豆 | 中国山西 | |
| F0002275 | 浅红菜豆 | 中国甘肃 | F0000049 | 架连豆 | 中国山西 | |
| F0002276 | 红菜豆 | 中国甘肃 | F0000054 | 坚豆 | 中国山西 | |
| F0002277 | 红黑花豆 | 中国甘肃 | F0000058 | 粘豆 | 中国山西 | |
| F0003342 | 白菜豆 | 中国甘肃 | F0000089 | 花菜豆 | 中国山西 | |
| F0004027 | 褐菜豆 | 中国甘肃 | F0000103 | 花灰灰豆 | 中国山西 | |
| F0005253 | 红腰豆 | 中国甘肃 | F0001292 | 洋菜豆 | 中国山西 | |
| F0005263 | 四季豆 | 中国甘肃 | F0001295 | 白眉豆 | 中国山西 | |
| F0005295 | 文昌四季豆 | 中国甘肃 | F0001318 | 白菜豆 | 中国山西 | |
| F0001802 | 白桩桩豆 | 中国贵州 | F0001339 | 白菜豆 | 中国山西 | |
| F0001809 | 玉滑豆 | 中国贵州 | F0001342 | 白粒菜豆 | 中国山西 | |
| F0001823 | 大红金豆 | 中国贵州 | F0001346 | 粉红紫皮连豆 | 中国山西 | |
| F0001850 | 木梳豆 | 中国贵州 | F0001361 | 红梅豆 | 中国山西 | |
| F0001914 | 灰褐子 | 中国贵州 | F0001380 | 小红豆 | 中国山西 | |
| F0002870 | 白云豆 | 中国贵州 | F0001394 | 紫红豆 | 中国山西 | |
| F0002972 | 黑籽鳝豆 | 中国贵州 | F0001416 | 红菜豆 | 中国山西 | |
| F0002974 | 黑金豆 | 中国贵州 | F0001428 | 紫连豆 | 中国山西 | |
| F0003010 | 笔划豆 | 中国贵州 | F0001434 | 黄红豆 | 中国山西 | |
| F0003067 | 白洋豆 | 中国贵州 | F0001445 | 黄红豆 | 中国山西 | |
| F0003082 | 硬壳鸡腰豆 | 中国贵州 | F0001459 | 无丝豆角 | 中国山西 | |
| F0003096 | 红芸豆 | 中国贵州 | F0001463 | 到底青 | 中国山西 | |
| F0003274 | 五花大铁悫豆 | 中国贵州 | F0001474 | 小灰豆 | 中国山西 | |
| F0003711 | 白金豆 | 中国贵州 | F0001497 | 小灰豆 | 中国山西 | |
| F0003740 | 四季豆 | 中国贵州 | F0001515 | 洋菜豆 | 中国山西 | |
| F0003757 | 大红豆 | 中国贵州 | F0001523 | 灰菜豆 | 中国山西 | |
| F0003758 | 四季豆 | 中国贵州 | F0001542 | 黑红豆 | 中国山西 | |
| F0003817 | 棒豆 | 中国贵州 | F0001567 | 黑粘菜豆 | 中国山西 | |
| F0003843 | 四季豆 | 中国贵州 | F0001600 | 白脸豆 | 中国山西 | |
| F0003970 | 黑花豆 | 中国贵州 | F0001619 | 粉花梅豆 | 中国山西 | |
| F0003990 | 大蛋豆 | 中国贵州 | F0001652 | 花色豆 | 中国山西 | |
| F0003992 | 红花豆 | 中国贵州 | F0001663 | 花刀豆 | 中国山西 | |
| F0003405 | 秋白芸豆 | 中国河北 | F0001670 | 梅豆 | 中国山西 | |
| F0003407 | 红芸豆 | 中国河北 | F0001675 | 麻雀蛋 | 中国山西 | |
| F0003453 | 黑芸豆 | 中国河北 | F0001691 | 花皮连豆 | 中国山西 | |
| F0005300 | 小黑芸豆 | 中国河北 | F0001704 | 花金豆 | 中国山西 | |
| F0005620 | 泰科六号 | 中国河北 | F0002223 | 洋菜豆 | 中国山西 | |
| F0000195 | 小白芸豆 | 中国黑龙江 | F0002229 | 桔黄梅豆 | 中国山西 | |
| F0000199 | 精米豆 | 中国黑龙江 | F0002240 | 小杂豆 | 中国山西 | |
| F0000203 | 精米豆 | 中国黑龙江 | F0002262 | 秋红豆 | 中国山西 | |
| F0000211 | 红芸豆 | 中国黑龙江 | F0001089 | 洋菜豆 | 中国陕西 | |
| F0000217 | 饭豆 | 中国黑龙江 | F0001090 | 白芸豆 | 中国陕西 | |
| F0000272 | 龙70-8014 | 中国黑龙江 | F0001102 | 洋蔓菜豆 | 中国陕西 | |
| F0000278 | 干枝密 | 中国黑龙江 | F0001200 | 花红豆 | 中国陕西 | |
| F0000281 | 白腰豆 | 中国黑龙江 | F0001244 | 没茎豆 | 中国陕西 | |
| F0000344 | 饭豆 | 中国黑龙江 | F0001924 | 洋小豆 | 中国陕西 | |
| F0000385 | 花饭豆 | 中国黑龙江 | F0001932 | 六十日早 | 中国陕西 | |
| F0000398 | 大马长 | 中国黑龙江 | F0001942 | 红豆 | 中国陕西 | |
| F0000404 | 兔子腿 | 中国黑龙江 | F0001972 | 五月黄 | 中国陕西 | |
| F0000415 | 花腰豆 | 中国黑龙江 | F0002032 | 四茬四季豆 | 中国陕西 | |
| F0000528 | 紫花豆 | 中国黑龙江 | F0002060 | 雀儿蛋 | 中国陕西 | |
| 统一编号Number | 种质Germplasm | 来源Origin | 统一编号Number | 种质Germplasm | 来源Origin | |
| F0001749 | 灰小豆 | 中国黑龙江 | F0002069 | 架四季豆 | 中国陕西 | |
| F0001757 | 毛毛豆 | 中国黑龙江 | F0002073 | 画眉豆 | 中国陕西 | |
| F0001760 | 兔脚 | 中国黑龙江 | F0002080 | 雀儿蛋 | 中国陕西 | |
| F0002503 | 白芸豆 | 中国黑龙江 | F0005090 | 大豆 | 中国陕西 | |
| F0002509 | 白芸豆 | 中国黑龙江 | F0005096 | 白豆 | 中国陕西 | |
| F0002521 | 黑芸豆 | 中国黑龙江 | F0005219 | 红芸豆 | 中国陕西 | |
| F0002523 | 黑芸豆 | 中国黑龙江 | F0002584 | 白四季豆 | 中国四川 | |
| F0002528 | 花芸豆 | 中国黑龙江 | F0002608 | 鸡腰子豆 | 中国四川 | |
| F0002535 | 大马掌 | 中国黑龙江 | F0002612 | 小猪腰豆 | 中国四川 | |
| F0002537 | 菜豆 | 中国黑龙江 | F0002620 | 红腰子豆 | 中国四川 | |
| F0003346 | 龙饭豆一号 | 中国黑龙江 | F0002623 | 红田季豆 | 中国四川 | |
| F0004837 | 黑芸豆 | 中国黑龙江 | F0002624 | 紫红豆 | 中国四川 | |
| F0004845 | 黑芸豆 | 中国黑龙江 | F0002628 | 腰子豆 | 中国四川 | |
| F0004851 | 黑芸豆 | 中国黑龙江 | F0002673 | 乌褐豆 | 中国四川 | |
| F0004867 | 奶花芸豆 | 中国黑龙江 | F0002683 | 硬壳豆 | 中国四川 | |
| F0004868 | 奶花芸豆 | 中国黑龙江 | F0002755 | 小黑四季豆 | 中国四川 | |
| F0004869 | 奶花芸豆 | 中国黑龙江 | F0002810 | 黑纹花豆 | 中国四川 | |
| F0004882 | 奶花芸豆 | 中国黑龙江 | F0002838 | 四十豆 | 中国四川 | |
| F0004883 | 奶花芸豆 | 中国黑龙江 | F0002863 | 奶花 | 中国四川 | |
| F0004886 | 奶花芸豆 | 中国黑龙江 | F0003673 | 花四季豆 | 中国四川 | |
| F0004890 | 奶花芸豆 | 中国黑龙江 | - | 圆奶花芸豆X | 中国新疆 | |
| F0004895 | 奶花芸豆 | 中国黑龙江 | F0000666 | 小杂豆 | 中国云南 | |
| F0004899 | 奶花芸豆 | 中国黑龙江 | F0000698 | 鸡腰子豆 | 中国云南 | |
| F0004910 | 奶花芸豆 | 中国黑龙江 | F0000867 | 菜豆 | 中国云南 | |
| F0004911 | 奶花芸豆 | 中国黑龙江 | F0000907 | 荷苞豆 | 中国云南 | |
| F0004923 | 奶花芸豆 | 中国黑龙江 | F0001006 | 雀蛋豆 | 中国云南 | |
| F0004926 | 奶花芸豆 | 中国黑龙江 | F0005027 | NV | 中国云南 | |
| F0005030 | 龙22-0579 | 中国黑龙江 | F0005031 | 云丰2号 | 中国云南 | |
| F0005033 | 品芸2号 | 中国黑龙江 | F0005035 | 260205 | 中国云南 | |
| F0005034 | 龙芸豆3号 | 中国黑龙江 | F0005039 | 260219 | 中国云南 | |
| F0005237 | 龙芸豆4号 | 中国黑龙江 | F0005361 | 浪台白菜豆 | 中国云南 | |
| F0005244 | 龙270267 | 中国黑龙江 | F0005363 | 哈施菜豆 | 中国云南 | |
| F0005245 | 龙270270 | 中国黑龙江 | F0005684 | 四季豆 | 中国云南 | |
| F0005246 | 龙270280 | 中国黑龙江 | F0005857 | 02A4 | 阿根廷 | |
| F0005248 | 龙270705 | 中国黑龙江 | F0005514 | GURGUTUBA | 巴西 | |
| F0005861 | 龙芸豆7号 | 中国黑龙江 | F0005524 | 60 dias | 巴西 | |
| - | 圆奶花芸豆H | 中国黑龙江 | F0005526 | Cubano | 巴西 | |
| F0003554 | 白刀豆 | 中国湖北 | F0005529 | Paraná 1 | 巴西 | |
| F0003567 | 黄眉豆 | 中国湖北 | F0005534 | Paraná | 巴西 | |
| F0003569 | 早菜豆 | 中国湖北 | F0005536 | BR IPA 10 | 巴西 | |
| F0003575 | 丛生四季豆 | 中国湖北 | F0005550 | Jalo Precoce | 巴西 | |
| F0003578 | 灰米米 | 中国湖北 | F0005555 | Novo Jalo | 巴西 | |
| F0003591 | 乌四季豆 | 中国湖北 | F0005568 | EMGOPA 201-Ouro | 巴西 | |
| F0003624 | 四月娥 | 中国湖北 | F0005574 | Carioca MG | 巴西 | |
| F0003626 | 小黑子 | 中国湖北 | F0005575 | Tambó | 巴西 | |
| F0003635 | 黑豆子 | 中国湖北 | F0005715 | 不详 | 巴西 | |
| F0003645 | 四季豆 | 中国湖北 | F0005716 | 不详 | 巴西 | |
| F0004523 | 花四季豆 | 中国湖北 | F0005717 | 不详 | 巴西 | |
| F0004529 | 白四季豆 | 中国湖北 | F0005718 | 不详 | 巴西 | |
| F0004538 | 草白豆 | 中国湖北 | F0005719 | 不详 | 巴西 | |
| F0004558 | 花饭豆 | 中国湖北 | F0005721 | 不详 | 巴西 | |
| F0004562 | 紫莲豆 | 中国湖北 | F0005875 | 不详 | 巴西 | |
| 统一编号Number | 种质Germplasm | 来源Origin | 统一编号Number | 种质Germplasm | 来源Origin | |
| F0004564 | 面豆角 | 中国湖北 | - | Manaus No5 | 巴西 | |
| F0004566 | 圆奶花芸豆 | 中国湖北 | - | Manaus No4 | 巴西 | |
| F0004568 | 紫轱辘坡豆 | 中国湖北 | - | Manaus No2 | 巴西 | |
| F0002305 | 豆 | 中国湖南 | - | Manaus No3 | 巴西 | |
| F0000440 | 精米豆 | 中国吉林 | F0002177 | 不详 | 法国 | |
| F0000442 | 白乌鸦菜豆 | 中国吉林 | F0003363 | 不详 | 法国 | |
| F0000444 | 白乌鸦菜豆 | 中国吉林 | F0003382 | 法引11号 | 法国 | |
| F0000445 | 白太 | 中国吉林 | F0003386 | 歌47 | 法国 | |
| F0000446 | 白太 | 中国吉林 | F0002125 | A48 | 哥伦比亚 | |
| F0000448 | 白芸豆 | 中国吉林 | F0002127 | W126 | 哥伦比亚 | |
| F0000452 | 饭豆 | 中国吉林 | F0002157 | BAT896 | 哥伦比亚 | |
| F0000460 | 白饭豆 | 中国吉林 | F0002166 | BAT331 | 哥伦比亚 | |
| F0000470 | 白饭豆 | 中国吉林 | F0002167 | BAT85 | 哥伦比亚 | |
| F0000473 | 红芸豆 | 中国吉林 | F0002169 | BAT336 | 哥伦比亚 | |
| F0000474 | 菜豆 | 中国吉林 | F0002170 | M101 | 哥伦比亚 | |
| F0000475 | 菜豆 | 中国吉林 | F0002171 | A51 | 哥伦比亚 | |
| F0000490 | 饭豆 | 中国吉林 | F0005767 | SER4 | 哥伦比亚 | |
| F0000492 | 罗唐豆 | 中国吉林 | F0005769 | SER6 | 哥伦比亚 | |
| F0000493 | 乌鸦菜豆 | 中国吉林 | F0005771 | SER8 | 哥伦比亚 | |
| F0000494 | 乌鸦菜豆 | 中国吉林 | F0005798 | SER35 | 哥伦比亚 | |
| F0000495 | 乌鸦菜豆 | 中国吉林 | F0005813 | SEN13 | 哥伦比亚 | |
| F0000501 | 饭大豆 | 中国吉林 | F0005830 | SEN32 | 哥伦比亚 | |
| F0000502 | 饭豆 | 中国吉林 | F0005833 | SEC1 | 哥伦比亚 | |
| F0000503 | 无季豆 | 中国吉林 | F0005841 | SEC10 | 哥伦比亚 | |
| F0002477 | 小白豆 | 中国吉林 | F0005843 | SEC14 | 哥伦比亚 | |
| F0002481 | 扁白豆 | 中国吉林 | F0005849 | 引红4 | 哥伦比亚 | |
| F0002486 | 白豆 | 中国吉林 | F0005879 | 不详 | 哥伦比亚 | |
| F0002489 | 白饭豆 | 中国吉林 | F0005910 | 不详 | 哥伦比亚 | |
| F0002492 | 红芸豆 | 中国吉林 | F0003370 | 不详 | 美国 | |
| F0002493 | 红芸豆 | 中国吉林 | - | BAT-93 | 美国 | |
| F0002502 | 奶花芸豆 | 中国吉林 | - | PI 633451 | 美国 | |
| - | 小红芸豆 | 中国吉林 | F0005919 | 不详 | 秘鲁 | |
| F0000117 | 天鹅豆 | 中国内蒙古 | F0005722 | 不详 | 墨西哥 | |
| F0000120 | 黄芸豆 | 中国内蒙古 | F0005924 | 不详 | 墨西哥 | |
| F0000126 | 挑花枚白连豆 | 中国内蒙古 | F0005093 | 芸豆 | 中国内蒙古 | |
| F0000143 | 饭豆 | 中国内蒙古 | F0005877 | 不详 | 委内瑞拉 | |
| F0000153 | 小菜豆 | 中国内蒙古 | F0004313 | 英国红芸豆 | 英国 | |
| F0000154 | 跃进豆 | 中国内蒙古 | F0004321 | ANT49 | CIAT | |
| F0001723 | 紫芸豆 | 中国内蒙古 | F0004322 | BRB-130 | CIAT | |
| F0001731 | 粉老来少 | 中国内蒙古 | F0004333 | UNS-27342-51 | CIAT | |
| F0001744 | 粉老来少 | 中国内蒙古 | F0004334 | LRK32 | CIAT | |
| F0002380 | 羊眼圈 | 中国内蒙古 | F0004339 | SEQ1006 | CIAT | |
| F0002399 | 红芸豆 | 中国内蒙古 | F0004341 | 9249-3 | CIAT | |
| F0002400 | 面豆荚 | 中国内蒙古 | F0004348 | FOI10 | CIAT | |
| F0002441 | 大红菜豆 | 中国内蒙古 | F0004349 | FOI11 | CIAT | |
| F0002462 | 花菜豆 | 中国内蒙古 | F0004350 | MCD2409 | CIAT | |
| F0002471 | 家雀蛋 | 中国内蒙古 | F0004357 | DOR483 | CIAT | |
| F0003497 | 架菜豆 | 中国内蒙古 | F0004374 | DRK139 | CIAT | |
| F0003503 | 白连豆 | 中国内蒙古 | F0004378 | FOT32 | CIAT | |
| F0004188 | 江米豆 | 中国内蒙古 | F0004395 | ISB-82-865 | CIAT | |
| F0004216 | 架菜豆 | 中国内蒙古 | F0004396 | DRK134 | CIAT | |
| F0004227 | 芸豆 | 中国内蒙古 | F0004398 | FOT25 | CIAT | |
| 统一编号Number | 种质Germplasm | 来源Origin | 统一编号Number | 种质Germplasm | 来源Origin | |
| F0004268 | 小芸豆 | 中国内蒙古 | F0004403 | DOR476 | CIAT | |
| F0004273 | 矮生红 | 中国内蒙古 | F0004404 | DOR482 | CIAT | |
| F0004587 | 紫轱辘坡豆 | 中国内蒙古 | F0004410 | VIVA | CIAT | |
| F0004592 | 奶花芸豆 | 中国内蒙古 | F0004413 | BAT58 | CIAT | |
| F0004594 | 金豆角 | 中国内蒙古 | F0002152 | R-5550 | 不详 | |
| F0005624 | 改良绿丰 | 中国山东 | F0002153 | R5350 | 不详 | |
| F0005629 | 四季架豆王 | 中国山东 | - | 圆奶花芸豆D | 不详 |
Note: Number is Seed Preservation Number of National Gene Bank. CIAT is International Centre for Tropical Agriculture
注:“统一编号”为国家种质库存种编号。“CIAT”为国际热带农业研究中心
1.2 试验方法
试验于2017-2018年进行。参考Liao等[18]的方法。幼苗的培养采用生长袋纸培系统,生长袋(30cm×25cm)购买于北京启维益成科技有限公司。首先利用发芽盒作为苗床进行种子萌发,培养3d左右,选取胚根长度为(2±0.10)cm的发芽种子转移至生长袋,向生长袋中加入100mL的蒸馏水,置于培养箱中培养,条件设置为:光照12h,25℃;黑暗12h,22℃。6d后将根系完整取出,利用CI-600(CID Inc,美国)根系扫描系统将根系扫描成图像,采用WinRHIZO Tron MF图像分析软件(Regent Instruments Inc,加拿大)[19]和直接测量方法获取根系表型数据。
1.3 测量指标及测量方法
参照Asfaw等[9]的方法测定各指标。每个品种取6株幼苗进行表型调查,其中总根长、根表面积、平均根直径、根体积通过图像分析系统获取。主根长、根干重、侧根数通过直接测量获取。主根长即下胚轴节到主根根尖的长度;从下胚轴节剪下根系,105℃杀青30min,60℃烘箱中烘烤48h,称量根干重。侧根总长=总根长-主根长,比根长=根干重/总根长。
1.4 基因组DNA的提取及基因型鉴定
1.5 数据分析
采用Excel 2007对所有数据进行整理,采用SPSS 19.0进行表型数据分析。利用PowerMarker V3.25软件[23]对基因型遗传多样性进行分析。采用Structure 2.3.4软件[24]分析群体结构,并获得最佳K值对应的Q矩阵,相关参数设置如下:K=1~10,重复15次,模拟参数迭代(length of burn-in period)设置为10000,蒙特卡罗迭代(markov chain monte carlo,MCMC)设置为100000,结果根据Evanno等[24]的方法通过ΔK判断群体结构的K值。采用SPAGeDi软件对基因型数据处理获得K矩阵[25]。采用Tassel 2.1软件的混合线性模型(mixed linear model,MLM)进行性状和标记之间的关联分析[26]。以P<0.01作为筛选显著位点的阈值。
2 结果与分析
2.1 表型数据分析
表2 普通菜豆幼苗期根系性状表型的统计分析
Table 2
| 性状Trait | 平均值Mean | 最小值Min. | 最大值Max. | 标准差Standard deviation | 变异系数Coefficient of variation (%) |
|---|---|---|---|---|---|
| 主根长Taproot length (cm) | 20.49 | 12.80 | 28.45 | 2.85 | 13.92 |
| 根干重Root dry weight (mg) | 29.56 | 10.00 | 75.00 | 10.94 | 37.03 |
| 侧根数Lateral root number | 12.05 | 5.40 | 22.80 | 3.18 | 26.38 |
| 总根长Total root length (cm) | 204.93 | 68.93 | 476.26 | 67.36 | 32.87 |
| 根表面积Root surface area (cm2) | 35.69 | 11.44 | 74.49 | 11.31 | 31.69 |
| 平均根直径Average root diameter (mm) | 0.56 | 0.26 | 0.77 | 0.06 | 10.09 |
| 根体积Root volume (cm3) | 0.50 | 0.15 | 1.04 | 0.16 | 33.08 |
| 侧根总长Lateral root length (cm) | 184.44 | 55.29 | 452.41 | 65.48 | 35.50 |
| 比根长Special root length (mg/cm) | 14.71 | 0.07 | 0.45 | 0.04 | 28.96 |
图1
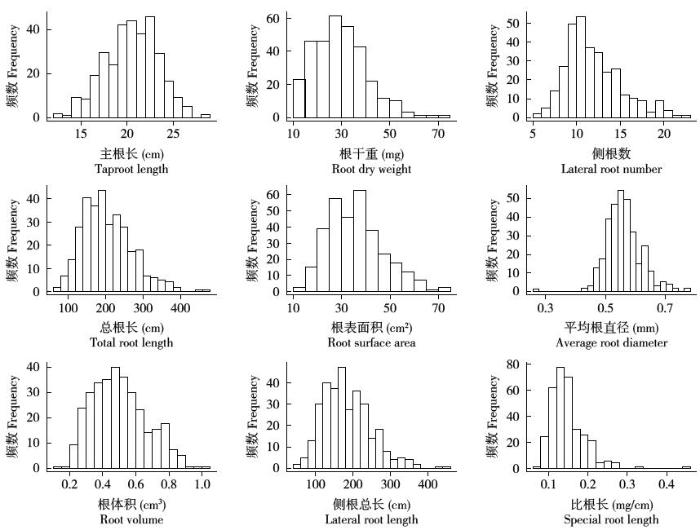
图1
普通菜豆幼苗期根系性状的频数分布
Fig.1
Frequency distribution of root-trait of common bean at seedling stage
2.2 表型相关性分析
对普通菜豆各根系性状表型数据进行相关性分析(表3)。其中平均根直径和比根长分别与主根长、侧根数、根表面积的相关性不显著,其余各性状两两之间的相关性均达到了极显著水平。其中侧根总长与总根长的相关系数(1.00)最大;比根长与根体积的相关系数(0.16)最小。平均根直径与总根长和侧根总长呈极显著负相关,相关系数分别为-0.22和-0.22,比根长与总根长和侧根总长呈极显著负相关,相关系数分别为-0.21和-0.21。
表3 根系性状的相关性
Table 3
| 性状 Trait | 主根长 Taproot length | 根干重 Root dry weight | 侧根数 Lateral root number | 总根长 Total root length | 根表面积 Root surface area | 平均根直径 Average root diameter | 根体积 Root volume | 侧根总长 Lateral root length | 比根长 Special root length |
|---|---|---|---|---|---|---|---|---|---|
| 主根长 Taproot length | -1 | ||||||||
| 根干重 Root dry weight | -0.50** | 1 | |||||||
| 侧根数 Lateral root number | -0.49** | 0.60** | -1 | ||||||
| 总根长 Total root length | -0.67** | 0.68** | -0.75** | -1 | |||||
| 根表面积 Root surface area | -0.66** | 0.79** | -0.76** | -0.96** | -1 | ||||
| 平均根直径 Average root diameter | -0.08 | 0.27** | -0.06 | -0.22** | -0.05 | -1 | |||
| 根体积 Root volume | -0.60** | 0.83** | -0.70** | -0.83** | -0.96** | -0.32** | 1 | ||
| 侧根总长 Lateral root length | -0.65** | 0.68** | -0.75** | -1.00** | -0.96** | -0.22** | 0.83** | -1 | |
| 比根长 Special root length | -0.07 | 0.54** | -0.05 | -0.21** | -0.03 | -0.62** | 0.16** | -0.21** | 1 |
Note: "**" indicates significant correlation at 0.01 level
注:“**”表示在0.01水平显著相关
2.3 基因型数据分析
利用均匀分布在11条染色体上的116个多态性SSR标记对324份材料进行基因分型分析,共检测到919个等位变异位点,等位变异位点的范围为2~27,平均每个标记检测到7.92个;利用PowerMarker软件对基因型数据进行分析,结果表明116个标记的基因多样性指数变动幅度介于0.0051~0.9098,每个标记的平均基因多样性指数为0.59,其中基因多样性指数大于平均数的标记有66个,占标记数的56.90%;多态性信息含量(polymorphism information content,PIC)的变动幅度介于0.0051~0.9033,平均值为0.54,其中PIC值大于平均数的标记有60个,占标记数的51.72%。结果表明这些SSR标记具有较高的基因多样性。
2.4 群体结构分析
通过STRUCTURE2.3.4软件对供试材料进行群体结构分析(图2),当K=2时,ΔK出现峰值,将供试材料分为两个亚群。这一结果与普通菜豆起源于两个基因库——安第斯基因库和中美基因库相符。将Q值大于0.70作为划分亚群的原则,将材料分为中美基因库群体和安第斯基因库群体,分别包含186和123份材料,分别占总材料数的57.41%和37.96%。剩余15份材料的两个Q值均小于0.70,将其单独划为一个混合群,占总材料数的4.63%。将各材料对应的Q值作为协变量用于SSR标记和表型性状的关联分析。
图2

图2
324份材料基于SSR标记的群体结构分析的K和ΔK的变化图
Fig.2
K and ΔK of population structure analysis based on SSR marker for 324 common bean accessions
2.5 根系性状关联分析
结合116个SSR标记和9个根系表型数据,利用Tassel 2.1软件中的MLM(Q+K)模型进行关联分析,以P<0.01为阈值筛选显著位点(表4),共检测到48个显著位点,表型贡献率范围为1.44%~16.09%,平均为5.18%。除主根长外,其余8个性状均检测到显著位点,其中平均根直径检测到的位点最多,为19个,表型贡献率为2.43%~9.55%;检测到与侧根数相关的位点仅有1个,表型贡献率为3.85%;检测到10个与根干重相关的位点,表型贡献率为1.44%~6.69%;检测到与总根长、根表面积、根体积、侧根总长和比根长关联的位点个数分别是3、3、4、3和5个,表型贡献率分别是4.63%~6.38%、2.17%~6.14%、2.28%~5.20%、4.77%~6.56%和4.81%~16.09%。
表4 基于混合模型的关联分析结果
Table 4
| 性状Trait | 标记位点 Locus | P值 P-value | 表型贡献率 R2 (%) |
|---|---|---|---|
| 根干重Root dry weight | CBS206 | 6.30×10-6 | 6.96 |
| CBS419 | 2.20×10-4 | 3.36 | |
| CBS110 | 5.67×10-4 | 3.83 | |
| P7S53 | 9.70×10-4 | 1.97 | |
| P7S191 | 0.0012 | 2.93 | |
| CBS139 | 0.0013 | 2.85 | |
| CBS261 | 0.0014 | 1.86 | |
| CBS208 | 0.0050 | 2.24 | |
| CBS345 | 0.0050 | 2.36 | |
| CBS307 | 0.0075 | 1.44 | |
| 侧根数Lateral root number | CBS28 | 0.0034 | 3.85 |
| 总根长Total root length | CBS408 | 8.50×10-5 | 6.38 |
| CBS22 | 0.0033 | 4.63 | |
| CBS250 | 0.0037 | 5.31 | |
| 根表面积Root surface area | CBS408 | 5.70×10-6 | 6.14 |
| P7S53 | 0.0055 | 2.17 | |
| CBS162 | 0.0090 | 4.46 | |
| 性状Trait | 标记位点 Locus | P值 P-value | 表型贡献率 R2 (%) |
| 平均根直径Average root diameter | CBS261 | 9.35×10-7 | 6.24 |
| CBS179 | 1.59×10-6 | 6.37 | |
| CBS43 | 2.18×10-6 | 6.33 | |
| CBS296 | 6.58×10-6 | 7.54 | |
| CBS51 | 7.00×10-6 | 8.07 | |
| CBS216 | 1.74×10-5 | 6.15 | |
| P11S130 | 6.62×10-5 | 7.38 | |
| CBS56 | 1.13×10-4 | 5.97 | |
| CBS28 | 2.37×10-4 | 5.18 | |
| CBS343 | 3.55×10-4 | 5.86 | |
| CBS82 | 5.18×10-4 | 9.55 | |
| CBS126 | 8.54×10-4 | 3.86 | |
| CBS211 | 0.0026 | 3.73 | |
| CBS229 | 0.0028 | 3.58 | |
| CBS349 | 0.0030 | 4.27 | |
| CBS381 | 0.0032 | 6.46 | |
| CBS286 | 0.0050 | 4.11 | |
| P11S186 | 0.0062 | 2.43 | |
| CBS248 | 0.0078 | 3.73 | |
| 根体积Root volume | CBS408 | 9.74×10-6 | 5.17 |
| CBS381 | 4.25×10-4 | 5.20 | |
| P7S53 | 0.0020 | 2.28 | |
| CBS162 | 0.0054 | 4.24 | |
| 侧根总长Lateral root length | CBS408 | 6.39×10-5 | 6.56 |
| CBS22 | 0.0028 | 4.77 | |
| CBS250 | 0.0043 | 5.30 | |
| 比根长Special root length | CBS206 | 1.13×10-10 | 16.09 |
| CBS419 | 2.23×10-7 | 7.35 | |
| CBS162 | 1.04×10-6 | 9.20 | |
| CBS149 | 1.37×10-4 | 6.07 | |
| CBS323 | 0.0019 | 4.81 |
表5 与已报道的根系相关QTL的比较结果
Table 5
3 讨论
根系是植物的三大营养器官之一,具有吸收养分和水分、物质同化和转化、固定作物等功能[28]。相对于地上部性状,根系性状的测量费时费力,故能够准确获取作物根系表型的手段和方法一直是作物根系研究的热点。根系性状的鉴定主要有室内测量和田间测量两类。室内测量的主要方法有水培法、凝胶室法和生长袋纸培法等[18,29-31],即将幼苗分别置于营养液、琼脂、湿纸的表面生长,在室内进行根系测量,可以严格控制根系生长环境,无伤获取根系,但只适用于幼苗期的根系。在普通菜豆中,苗期根系的研究多用生长袋纸培法[18,32]。廖红等[32]采用营养袋纸培法研究普通菜豆根系的生长,发现移苗6d后的根系各性状与砂培系统中28d的根系各性状显著相关,说明营养袋纸培法是一种简便可行的研究根系方法。本研究利用生长袋纸培法对不同菜豆种质进行鉴定,发现根干重、侧根数、总根长、根表面积等性状均呈现较大的变异,不同的根性状之间存在不同程度的相关性。
关联分析是检测数量性状相关位点或基因的重要手段,而群体结构是影响关联分析的主要因素。群体结构会造成关联分析结果的假阳性,即检测的显著位点不是由于表型性状和功能位点的关联而造成的,而是由群体结构所导致的[33]。有研究表明,相较只考虑群体结构(Q矩阵)或亲缘关系(K矩阵)的GLM(Q)和MLM(K)模型,将Q矩阵和K矩阵作为协变量的MLM(Q+K)模型能够更好消除伪关联[34]。因此,本研究利用STRCTURE软件进行群体结构分析,并获取Q矩阵作为协变量进行MLM模型的关联分析。当供试材料等位变异平均频率特征类型数K=2时,ΔK达到峰值,即将供试群体分成2个亚群,这与前人通过形态标记、同工酶、朊蛋白、分子标记等证实普通菜豆起源于中美基因库和安第斯基因库的结果一致[35,36]。
本研究对324份普通菜豆材料的9个根系性状进行了关联分析,为有效减少由群体结构与亲缘关系引起的假阳性,将阈值设置为P<0.01,共检测到48个显著位点。其中,10个一因多效位点中,7个位点分别与两个性状相关联;2个位点分别与3个性状相关联。位点P7S53与根干重、根表面积和根体积相关联,位点CBS162与根表面积、根体积和比根长相关联;位点CBS408与总根长、根表面积、根体积和侧根总长相关联,说明这些标记有助于对多个性状进行改良。前人对普通菜豆的根系研究表明,根系与耐旱性[9]、磷获取[10,27,37]、耐铝性[11]等性状有关。本研究中发现有5个位点与前人检测到的根系QTL位置相符。例如,Ochoa等[27]在不同磷环境下对普通菜豆的根系进行遗传研究,检测到20个根系相关的QTL,其中4个与比根长相关的QTL分别与本研究中的显著位点CBS56、CBS208、CBS211、CBS286相符。Lopez-Marin等[11]在铝环境下对普通菜豆根系进行研究,检测到5个与根干重相关的QTL,其中1个QTL与本研究中的显著位点P11S186相符。虽然对应的性状不同,但这些性状在本研究中都呈显著正相关。其余的43个位点可能是新检测到的与根系关联的标记,这些标记为进一步理解普通菜豆根系性状的遗传机理提供理论参考,也为分子标记辅助选择改良普通菜豆根系奠定了基础。
参考文献
Root adaptations to soils with low fertility and aluminium toxicity
DOI:10.1093/aob/mcw073
URL
PMID:5055624
[本文引用: 1]

BackgroundPlants depend on their root systems to acquire the water and nutrients necessary for their survival in nature, and for their yield and nutritional quality in agriculture. Root systems are complex and a variety of root phenes have been identified as contributors to adaptation to soils with low fertility and aluminium (Al) toxicity. Phenotypic characterization of root adaptations to infertile soils is enabling plant breeders to develop improved cultivars that not only yield more, but also contribute to yield stability and nutritional security in the face of climate variability. ScopeIn this review the adaptive responses of root systems to soils with low fertility and Al toxicity are described. After a brief introduction, the purpose and focus of the review are outlined. This is followed by a description of the adaptive responses of roots to low supply of mineral nutrients [with an emphasis on low availability of nitrogen (N) and phosphorus (P) and on toxic levels of Al]. We describe progress in developing germplasm adapted to soils with low fertility or Al toxicity using selected examples from ongoing breeding programmes on food (maize, common bean) and forage/feed (Brachiariaspp.) crops. A number of root architectural, morphological, anatomical and metabolic phenes contribute to the superior performance and yield on soils with low fertility and Al toxicity. Major advances have been made in identifying root phenes in improving adaptation to low N (maize), low P (common bean) or high Al [maize, common bean, species and hybrids of brachiariagrass, bulbous canarygrass (Phalaris aquatica) and lucerne (Medicago sativa)]. ConclusionsAdvanced root phenotyping tools will allow dissection of root responses into specific root phenes that will aid both conventional and molecular breeders to develop superior cultivars. These new cultivars will play a key role in sustainable intensification of crop ivestock systems, particularly in smallholder systems of the tropics. Development of these new cultivars adapted to soils with low fertility and Al toxicity is needed to improve global food and nutritional security and environmental sustainability.
Regulation of plant root system architecture:implications for crop advancement
DOI:10.1016/j.copbio.2014.11.015
URL
PMID:25448235
[本文引用: 2]
Root system architecture (RSA) plays a major role in plant fitness, crop performance, and grain yield yet only recently has this role been appreciated. RSA describes the spatial arrangement of root tissue within the soil and is therefore crucial to nutrient and water uptake. Recent studies have identified many of the genetic and environmental factors influencing root growth that contribute to RSA. Some of the identified genes have the potential to limit crop loss caused by environmental extremes and are currently being used to confer drought tolerance. It is hypothesized that manipulating these and other genes that influence RSA will be pivotal for future crop advancements worldwide.
QTL for root angle and number in a population developed from bread wheats (Triticum aestivum) with contrasting adaptation to water-limited environments
DOI:10.1007/s00122-013-2074-0
URL
PMID:23525632
[本文引用: 1]
AbstractRoot architecture traits in wheat are important in deep soil moisture acquisition and may be used to improve adaptation to water-limited environments. The genetic architecture of two root traits, seminal root angle and seminal root number, were investigated using a doubled haploid population derived from SeriM82 and Hartog. Multiple novel quantitative trait loci (QTL) were identified, each one having a modest effect. For seminal root angle, four QTL (61log() >3) were identified on 2A, 3D, 6A and 6B, and two suggestive QTL (61log() >2) on 5D and 6B. For root number, two QTL were identified on 4A and 6A with four suggestive QTL on 1B, 3A, 3B and 4A. QTL for root angle and root number did not co-locate. Transgressive segregation was found for both traits. Known major height and phenology loci appear to have little effect on root angle and number. Presence or absence of the T1BL.1RS translocation did not significantly influence root angle. Broad sense heritability () was estimated as 5002% for root angle and 3102% for root number. Root angle QTL were found to be segregating between wheat cultivars adapted to the target production region indicating potential to select for root angle in breeding programs.
LEAF TIP NECROSIS1 plays a pivotal role in the regulation of multiple phosphate starvation responses in rice
DOI:10.1104/pp.110.170209
URL
PMID:21317339
[本文引用: 1]
Abstract Although phosphate (Pi) starvation signaling is well studied in Arabidopsis (Arabidopsis thaliana), it is still largely unknown in rice (Oryza sativa). In this work, a rice leaf tip necrosis1 (ltn1) mutant was identified and characterized. Map-based cloning identified LTN1 as LOC_Os05g48390, the putative ortholog of Arabidopsis PHO2, which plays important roles in Pi starvation signaling. Analysis of transgenic plants harboring a LTN1 promoter:: -glucuronidase construct revealed that LTN1 was preferentially expressed in vascular tissues. The ltn1 mutant exhibited increased Pi uptake and translocation, which led to Pi overaccumulation in shoots. In association with enhanced Pi uptake and transport, some Pi transporters were up-regulated in the ltn1 mutant in the presence of sufficient Pi. Furthermore, the elongation of primary and adventitious roots was enhanced in the ltn1 mutant under Pi starvation, suggesting that LTN1 is involved in Pi-dependent root architecture alteration. Under Pi-sufficient conditions, typical Pi starvation responses such as stimulation of phosphatase and RNase activities, lipid composition alteration, nitrogen assimilation repression, and increased metal uptake were also activated in ltn1. Moreover, analysis of OsmiR399-overexpressing plants showed that LTN1 was down-regulated by OsmiR399. Our results strongly indicate that LTN1 is a crucial Pi starvation signaling component downstream of miR399 involved in the regulation of multiple Pi starvation responses in rice.
QTL mapping and epistasis analysis of brace root traits in maize
DOI:10.1007/s11032-011-9655-x
URL
[本文引用: 1]
Abstract21112 to affect the expression of TBRTN. Therefore, a complex network controlling the traits was found in maize. These results provide useful information for understanding the molecular mechanisms controlling root architecture.
Molecular mapping of the brace root traits in sorghum (Sorghum bicolor L. Moench)
DOI:10.1270/jsbbs.64.193
URL
PMID:4065327
[本文引用: 1]
Abstract The presence and morphology of plant brace roots are important root architecture traits. Brace roots contribute significantly to effective anchorage and water and nutrient uptake during late growth and development, and more importantly, have a substantial influence on grain yield under soil flooding or water limited conditions. However, little is known about the genetic mechanisms that underlie brace root traits. In this study, quantitative trait loci (QTLs) for presence of brace roots from the sorghum landrace "Sansui" were mapped and associated molecular markers were identified. A linkage map was constructed with 109 assigned simple sequence repeat markers using a F2 mapping population derived from the cross Sansui/Jiliang 2. Two QTLs associated with presence of brace roots were localized on chromosomes 6 and 7. The major QTL on chromosome 7 between markers Dsenhsbm7 and Xcup 70 explained about 52.5% of the phenotypic variation, and the minor QTL on chromosome 6 was flanked by Xtxp127 and Xtxp6 and accounted for 7.0% of phenotypic variation. These results will provide information for the improvement of sorghum root architecture associated with brace roots.
QTL for nodal root angle in sorghum (Sorghum bicolor L. Moench) co-locate with QTL for traits associated with drought adaptation
DOI:10.1007/s00122-011-1690-9
URL
PMID:21938475
[本文引用: 1]
Nodal root angle in sorghum influences vertical and horizontal root distribution in the soil profile and is thus relevant to drought adaptation. In this study, we report for the first time on the mapping of four QTL for nodal root angle ( qRA ) in sorghum, in addition to three QTL for root dry weight, two for shoot dry weight, and three for plant leaf area. Phenotyping was done at the six leaf stage for a mapping population ( n = 141) developed by crossing two inbred sorghum lines with contrasting root angle. Nodal root angle QTL explained 58.2% of the phenotypic variance and were validated across a range of diverse inbred lines. Three of the four nodal root angle QTL showed homology to previously identified root angle QTL in rice and maize, whereas all four QTL co-located with previously identified QTL for stay-green in sorghum. A putative association between nodal root angle QTL and grain yield was identified through single marker analysis on field testing data from a subset of the mapping population grown in hybrid combination with three different tester lines. Furthermore, a putative association between nodal root angle QTL and stay-green was identified using data sets from selected sorghum nested association mapping populations segregating for root angle. The identification of nodal root angle QTL presents new opportunities for improving drought adaptation mechanisms via molecular breeding to manipulate a trait for which selection has previously been very difficult.
Overexpression of a NAC transcription factor enhances rice drought and salt tolerance
DOI:10.1016/j.bbrc.2008.12.163
URL
PMID:19135985
[本文引用: 1]
The plant-specific NAC (NAM, ATAF1/2, CUC2) transcription factors play diverse roles in plant development and stress responses. In this study, a rice NAC gene, ONAC045, was functionally characterized, especially with regard to its role in abiotic stress resistance. Expression analysis revealed that ONAC045 was induced by drought, high salt, and low temperature stresses, and abscisic acid (ABA) treatment in leaves and roots. Transcriptional activation assay in yeast indicated that ONAC045 functioned as a transcriptional activator. Transient expression of GFP-ONAC045 in onion epidermal cells revealed that ONAC045 protein was localized in the nucleus. Transgenic rice plants overexpressing ONAC045 showed enhanced tolerance to drought and salt treatments. Two stress-responsive genes were upregulated in transgenic rice. Together, these results suggest that ONAC045 encodes a novel stress-responsive NAC transcription factor and is potential useful for engineering drought and salt tolerant rice.
Quantitative trait loci for rooting pattern traits of common beans grown under drought stress versus non-stress conditions
DOI:10.1007/s11032-011-9654-y
URL
[本文引用: 4]
AbstractDrought is the major abiotic constraint contributing to yield reduction in common bean (Phaseolus vulgaris L.) worldwide. An increasing scarcity of water in the future will make improving adaptation to drought stress a major objective of most crop breeding efforts. Drought avoidance by increased extraction of soil moisture from greater depth under drought conditions is an adaptive mechanism of common bean. A recombinant inbred line population of DOR364 BAT477 was evaluated for rooting pattern traits in soil cylinder tubes under soil drying (progressive water stress) and non-stress (well-watered with 80% of field capacity) treatments in a greenhouse. One of the parents, BAT 477, is a deep-rooting genotype while the other parent, DOR 364, is a commercial cultivar in Central America. The recombinant inbred line population expressed quantitative variation and transgressive segregation for ten rooting pattern traits as well as five shoot traits of 48-day-old plants. A mixed model quantitative trait locus (QTL) mapping analysis was carried out using a genetic map constructed with 165 genetic markers that covered 11 linkage groups of the common bean genome. Genotype estimates were calculated from best design and spatial effects model for each trait. A total of 15 putative QTL were identified for seven rooting pattern traits and four shoot traits. The QTL detected were scattered over five of the 11 linkage groups. The QTL detected for all the root traits except total root length and fine root length were main effect QTL and did not interact with the level of water supply. The total root length and fine root length QTL with significant QTL environment interaction only differed in magnitude of effect, and interaction was of a non-crossover type. Other QTL for total root length, fine roots, thick roots, root volume and root biomass were co-localized and also explained relatively more genetic variance. This suggests that the QTL affecting root traits in common beans are based on constitutive expression of genes and that drought avoidance based on deep rooting, longer root length, thicker roots, increasing root length distribution with depth, root volume and root biomass can be used in molecular breeding. The positive alleles for most of the QTL detected in this study were derived from the paternal parent BAT477. The results from the present analyses highlighted the feasibility of marker-aided selection as an alternative to conventional labor-intensive, phenotypic screening of drought avoidance root traits.
Quantitative trait loci for root architecture traits correlated with phosphorus acquisition in common bean
DOI:10.2135/cropsci2005.0226 URL [本文引用: 3]
Quantitative trait loci for root morphology traits under aluminum stress in common bean (Phaseolus vulgaris L.)
DOI:10.1007/s00122-009-1051-0
URL
PMID:19436988
[本文引用: 6]
Aluminum (Al) toxicity is a major limiting factor of crop production in acid soils, which are found mostly in developing countries of the tropics and sub-tropics. Common bean ( Phaseolus vulgaris L.) is particularly sensitive to Al toxicity; and development of genotypes with better root growth in Al-toxic soils is a priority. The objectives of the present study were to physiologically assess root architectural traits in a recombinant inbred line (RIL) population of common bean that contrasts for Al resistance (DOR36402×02G19833) and to identify quantitative trait loci (QTL) controlling root growth under two nutrient solutions, one with 2002μM Al concentration and the other without Al, both at pH 4.5. A total of 24 QTL were found through composite interval mapping analysis, 9 for traits under Al treatment, 8 for traits under control treatment, and 7 for relative traits. Root characteristics expressed under Al treatment were found to be under polygenic control, and some QTL were identified at the same location as QTL for tolerance to low phosphorous stress, thus, suggesting cross-links in genetic control of adaptation of common bean to different abiotic stresses.
Root phenotyping for drought tolerance in bean landraces from Calabria (Italy)
DOI:10.1111/jac.12124
URL
[本文引用: 1]
Abstract Common bean ( Phaseolus vulgaris ) is cultivated throughout Latin America and Africa, and for the European community, in Italy and Spain, areas are mainly subjected to drought stress which is predict to worsen by regional climatic models. The aims of this work were to identify the drought-tolerant and drought-sensitive bean landraces using drought tolerance and phenotypic plasticity indexes and to dissect the root morphological and 2D-architecture traits related to drought tolerance. Thirty-one landraces from diverse gene pools and areas of the Calabria region (South Italy), with different habits and morphological traits, were screened for drought tolerance in a hydroponic system. Root phenotyping was conducted by image analysis. Drought tolerance screening identified two landraces as drought tolerant and sensitive, respectively. Under drought stress, the drought-tolerant landrace exhibited several interesting root traits such as a higher root length, surface area and, above all, the fineness of the whole root systems and, with emphasis, of the higher order roots. Drought stress induced plastic root responses in both bean landraces but with contrasting patterns. The drought-tolerant landrace exhibited a dimorphic-rooted strategy, which could be included in future utility for bean breeding programmes in drought-prone environments.
Root traits and their potential links to plant ideotypes to improve drought resistance in common bean
DOI:10.1007/s40626-017-0090-1
URL
[本文引用: 1]
Drought stress limits growth and yield of crops, particularly under smallholder production systems with minimal use of inputs and edaphic limitations such as nitrogen (N) deficiency. The development of genotypes adapted to these conditions through genetic improvement is an important strategy to address this limitation. The identification of morpho-physiological traits associated with drought resistance contributes to increasing the efficiency of breeding programs. A set of 36 bean genotypes belonging to the Middle American gene pool was evaluated. A greenhouse study using soil cylinders was conducted to determine root vigor traits (total root length and fine root production) under drought stress. Two field trials were conducted to determinate grain yield, symbiotic nitrogen fixation (SNF) ability and other shoot traits under drought stress. Field data on grain yield and other shoot traits measured under drought were related with the greenhouse data on root traits under drought conditions to test the relationships between shoot traits and root traits. Response of root vigor to drought stress appeared to be related with ideotypes of water use (water savers and water spenders). The water spender ideotypes presented deeper root system, while the water saver ideotypes showed a relatively shallower root system. Increase in SNF ability under drought stress was associated with greater values of mean root diameter while greater acquisition of N from soil was associated with finer root system. We identified seven common bean lines (SEA 15, NCB 280, SCR 16, SMC 141, BFS 29, BFS 67 and SER 119) that showed greater root vigor under drought stress in the greenhouse and higher values of grain yield under drought stress in the field. These lines could serve as parents for improving drought resistance in common bean.
Assessing root traits associated with root rot resistance in common bean
DOI:10.1016/j.fcr.2003.08.001
URL
[本文引用: 1]
Detecting differences in root architecture and growth patterns among common bean ( Phaseolus vulgaris L.) genotypes may provide unique selection criteria for genetic resistance to Fusarium root rot. Genetic variation in root system architecture was quantified for 10 contrasting bean genotypes that represent four common bean classes (kidney, cranberry, black, and snap bean) under greenhouse conditions and under root rot disease pressure in the field. Genetic variation existed in root architecture among common bean classes and was highly significant under field conditions. Variation in root traits was minimal under environmentally controlled, greenhouse conditions. Results from the field evaluation suggest that a greater number of adventitious roots can contribute to root rot resistance, where the three most resistant genotypes accumulated large amounts of biomass in adventitious roots. In the field environment, total root system dry weight was correlated to fine ( r=0.74, P<0.001) and intermediate ( r=0.66, P<0.01) root classes. Plasticity of root system response was high, indicating the value of screening in the field environment.
Natural variation in common bean (Phaseolus vulgaris L.) for root traitsand biomass partitioning under drought
Development and distribution of root system in two grain sorghum cultivars originated from Sudan under drought stress
DOI:10.1626/pps.8.553
URL
[本文引用: 1]
The difference in rooting pattern between two grain sorghum cultivars differing in drought tolerance was investigated under drought stress. The cultivars, Gadambalia (drought-tolerant) and Tabat (drought-susceptible), were grown in bottomless wooden or acrylic root boxes to examine root parameters. Gadambalia consistently exhibited higher dry matter production and leaf water potential than Tabat under drought stress in both root boxes. In the experiment with wooden root boxes, under a drought condition, Gadambalia extracted more water from deep soil layers (1.1-1.5 m), which was estimated from the reduction in soil water content, than Tabat. This was because Gadambalia had a significantly higher root length density in these soil layers. The high root length density was due to enhanced lateral root development in Gadambalia. In the other experiment with acrylic root boxes, though total root length in the upper soil layer (0-0.5 m) was declined by limited irrigation in both cultivars, the reduction in Gadambalia was moderate compared with that in Tabat owing to the maintenance of fine root growth. Unlike Tabat, Gadambalia had an ability to produce the nodal roots from higher internodes even under drought, which resulted in the high nodal root length of Gadambalia. The growth angle of nodal roots was significantly correlated with root diameter, and the nodal roots from the higher internodes had large diameters and penetrated into the soil more vertically. These results indicate that the responses of roots (i.e. branching and/or growth of lateral root, and nodal root emergence from higher internodes) to soil dryness could be associated with the drought tolerance of Gadambalia.
大豆根系生长和活性特点的研究
本试验利用框架剖面法测定了大豆根系的生长动态和根系活性变化.结果表明:大豆根系生长过程呈S型曲线变化,形成慢生长(Ve-V_3),快速生长(V_3-R_5)和衰老(R_5),三个阶段,高峰值出现在 R_4一R_5阶段.根系活性变化与根系生长特点相似,R_1时期之前根系活性逐渐增强,R_2时期之后根系活性下降.根系活性变化比根系生长提前
Effect of phosphorus availability on basal root shallowness in common bean
DOI:10.1023/A:1010381919003
URL
PMID:11729851
[本文引用: 3]
Root gravitropism may be an important element of plant response to phosphorus availability because it determines root foraging in fertile topsoil horizons, and thereby phosphorus acquisition. In this study we seek to test this hypothesis in both two dimensional paper growth pouch and three-dimensional solid media of sand and soil cultures. Five common bean (Phaseolus vulgaris L.) genotypes with contrasting adaptation to low phosphorus availability were evaluated in growth pouches over 6 days of growth, and in sand culture and soil culture over 4 weeks of growth. In all three media, phosphorus availability regulated the gravitropic response of basal roots in a genotype-dependent manner. In pouches, sand, and soil, the phosphorus-inefficient genotype DOR 364 had deeper roots with phosphorus stress, whereas the phosphorus-efficient genotype G19833 responded to phosphorus stress by producing shallower roots. Genotypes were most responsive to phosphorus stress in sand culture, where relative root allocation to the 0-3- and 3-6-cm horizons increased 50% with phosphorus stress, and varied 300% (3-6 cm) to 500% (0-3 cm) among genotypes. Our results indicate that (1) phosphorus availability regulates root gravitropic growth in both paper and solid media, (2) responses observed in young seedlings continue throughout vegetative growth, (3) the response of root gravitropism to phosphorus availability varies among genotypes, and (4) genotypic adaptation to low phosphorus availability is correlated with the ability to allocate roots to shallow soil horizons under phosphorus stress.
EZ-Rhizo:integrated software for the fast and accurate measurement of root system architecture
DOI:10.1111/tpj.2009.57.issue-5 URL [本文引用: 1]
Increasing cotton genome coverage with polymorphic SSRs as revealed by SSCP
DOI:10.1139/g2012-032
URL
PMID:22670804
[本文引用: 1]
Simple sequence repeat (SSR) markers are widely used in plant genetics and breeding. However, there are many SSR markers that do not reveal polymorphism in . Traditional SSR genotyping methods only provide information on product sizes. This leaves many marker polymorphism undetected, thus, lowering the utility of SSRs. In the present study, monomorphic SSRs between two mapping parents, 'Emian22' and 3-79, were subjected to single-strand conformation polymorphism (SSCP) analysis to reveal polymorphism. Of the 4194 monomorphic SSR primer pairs, 158 pairs (3.77%) showed polymorphism and revealed 174 polymorphic loci. Sequence analysis showed that the differences in PCR products between the mapping parents were solely due to base transition or transversion, which was in agreement with SSCP principles. SSCP also revealed SSRs with motifs of AT/TA and GAA/CTT were more polymorphic in and trinucleotides, respectively. Genetic mapping integrated 160 loci into our interspecific BC(1) linkage map, 5 of which associated with QTLs related to fiber quality. The technique discussed in the present study enables us to detect polymorphism of monomorphic SSRs, and increase the utilization efficiency of the existing SSR primers.
Development of mapped simple sequence repeat markers from common bean (Phaseolus vulgaris L.) based on genome sequences of a Chinese landrace and diversity evaluation
DOI:10.1007/s11032-013-9949-2
URL
[本文引用: 1]
Microsatellite or single sequence repeat (SSR) markers have been commonly used in genetic research in many crop species, including common bean ( Phaseolus vulgaris L.). A limited number of existing SSR markers have been designed from high-throughput sequencing of the genome, warranting the exploitation of new SSR markers from genomic regions. In this paper, we sequenced total DNA from the genotype Hong Yundou with a 454-FLX pyrosequencer and found numerous SSR loci. Based on these, a large number of SSR markers were developed and 90 genomic-SSR markers with clear bands were tested for mapping and diversity detection. The new SSR markers proved to be highly polymorphic for molecular polymorphism, with an average polymorphism information content value of 0.44 in 131 Chinese genotypes and breeding lines, effective for distinguishing Andean and Mesoamerican genotypes. In addition, we integrated 85 primers of the 90 polymorphism markers into the bean map using an F2 segregating population derived from Hong Yundou crossed with Jingdou. The distribution of SSR markers among 11 chromosomes was not random and tended to cluster on the linkage map, with 14 new markers mapped on chromosome Pv01, whereas only four loci were located on chromosome Pv04. Overall, these new markers have potential for genetic mapping, genetic diversity studies and map-based cloning in common bean.
Identification of presumed ancestral dna sequences of phaseolin in phaseolus vulgaris
DOI:10.1073/pnas.92.4.1101 URL [本文引用: 1]
PowerMarker:an integrated analysis environment for genetic marker analysis
DOI:10.1093/bioinformatics/bti282
URL
PMID:15705655
[本文引用: 1]
PowerMarker delivers a data-driven, integrated analysis environment (IAE) for genetic data. The IAE integrates data management, analysis and visualization in a user-friendly graphical user interface. It accelerates the analysis lifecycle and enables users to maintain data integrity throughout the process. An ever-growing list of more than 50 different statistical analyses for genetic markers has been implemented in PowerMarker.
Detecting the number of clusters of individuals using the software STRUCTURE:a simulation study
DOI:10.1111/j.1365-294X.2005.02553.x
URL
PMID:15969739
[本文引用: 2]
The identification of genetically homogeneous groups of individuals is a long standing issue in population genetics. A recent Bayesian algorithm implemented in the software structure allows the identification of such groups. However, the ability of this algorithm to detect the true number of clusters ( K ) in a sample of individuals when patterns of dispersal among populations are not homogeneous has not been tested. The goal of this study is to carry out such tests, using various dispersal scenarios from data generated with an individual-based model. We found that in most cases the estimated 'log probability of data' does not provide a correct estimation of the number of clusters, K . However, using an ad hoc statistic K based on the rate of change in the log probability of data between successive K values, we found that structure accurately detects the uppermost hierarchical level of structure for the scenarios we tested. As might be expected, the results are sensitive to the type of genetic marker used (AFLP vs. microsatellite), the number of loci scored, the number of populations sampled, and the number of individuals typed in each sample.
SPAGEDi:a versatile computer program to analyse spatial genetic structure at the individual or population levels
DOI:10.1046/j.1471-8286.2002.00305.x URL [本文引用: 1]
TASSEL:software for association mapping of complex traits in diverse samples
DOI:10.1093/bioinformatics/btm308 URL [本文引用: 1]
QTL Analysis of adventitious root formation in common bean under contrasting phosphorus availability
DOI:10.2135/cropsci2005.12-0446
URL
[本文引用: 8]
Low phosphorus availability is a primary constraint to crop production in developing countries. Adventitious roots play an important role in phosphorus acquisition, as they are localized near the soil surface where phosphorus is relatively abundant. A population of recombinant inbred lines of Phaseolus valgatis L. (G2333/G19839) was screened under high- and low-phosphorus conditions in the greenhouse and field. We observed phenotypic variation and transgressive segregation for adventitious root traits in both environments. Allometric analysis revealed that although the taproot and basal roots are closely linked to shoot growth, recombinant inbred tine (RILs) differ substantially in biomass allocation for adventitious roots. A linkage map with 149 genetic markers and a total cumulative map length of 1175 cM was used to identify, a total of 19 QTL across 8 of the 11 linkage groups. Together these quantitative trait loci (QTL) accounted for 119 to 61% of the total phenotypic variation for adventitious root traits in the field and 18 to 39% under greenhouse conditions. Two major QTL for adventitious rooting under low phosphorus conditions in the field were observed on linkage groups B2 and B9 that together accounted for 61% of the observed phenotypic variation. We conclude that adventitious rooting under low phosphorus is a feasible target for bean breeding.
大豆幼苗根系性状的QTL分析
DOI:10.3724/SP.J.1006.2011.01151
URL
Magsci
[本文引用: 1]
为研究大豆幼苗期根系性状的遗传规律,以中豆29和中豆32构建的RIL群体为材料,在V2期测定水培幼苗根系性状(主根长、侧根数、根重、根体积和根冠比等)及相关性状(株重、茎叶重和下胚轴重等),以方差分析方法估算遗传参数,并采用复合区间作图法对大豆幼苗期根系等性状进行QTL定位。结果表明,在8个染色体上检测到20个根系及相关性状QTL,其中9个主效QTL位于第11和第14染色体,表型贡献率在10.5%~26.1%之间。在第11和第14染色体上,部分根系性状QTL与地上部性状QTL处于同一位置,其QTL的共位性与形态性状表型相关分析结果一致,反映了根系性状与地上部性状存在一定的关联。
3D reconstruction and dynamic modeling of root architecture in situ and its application to crop phosphorus research
DOI:10.1111/tpj.2009.60.issue-6 URL [本文引用: 1]
Growth of axile and lateral roots of maize:I development of a phenotying platform
DOI:10.1007/s11104-009-9984-2
URL
The objective of this study was to develop a phenotyping platform for the non-destructive, digital measurement of early root growth of axile and lateral roots and to evaluate its suitability for identifying maize ( Zea mays L.) genotypes with contrasting root development. The system was designed to capture images of the root system within minutes and to batch process them automatically. For system establishment, roots of the inbred line Ac7729/TZSRW were grown until nine days after germination on the surface of a blotting paper in pouches. An A4 scanner was used for image acquisition followed by digital image analysis. Image processing was optimized to enhance the separation between the roots and the background and to remove image noise. Based on the root length in diameter-class distribution (RLDD), small-diameter lateral roots and large-diameter axile roots were separated. Root systems were scanned daily to model the growth dynamics of these root types. While the axile roots exhibited an almost linear growth, total lateral root length increased exponentially. Given the determined exponential growth, it was demonstrated that two plants, germinated one day apart but with the same growth rates differed in root length by 100%. From the growth rates we were able to identify contrasting genotypes from 236 recombinant inbred lines (RILs) of the CML444 x SC-Malawi cross. Differences in the growth of lateral roots of two selected RILs were due to differences in the final length and linear density of the primary lateral roots, as proven by the manual reanalysis of the digital images. The high throughput makes the phenotyping platform attractive for routine genetic studies and other screening purposes.
Imaging and analysis platform for automatic phenotyping and trait ranking of plant root systems
DOI:10.1104/pp.109.150748 URL [本文引用: 1]
菜豆根构型对低磷胁迫的适应性变化及基因型差异
DOI:10.3321/j.issn:1672-9072.2000.02.009
URL
[本文引用: 2]
利用特殊设计的营养袋纸培和分层式磷控释砂培等根系生长系统结合计算机图像分析技术 ,以基根根长在生长介质各层的相对分布和基根平均生长角度为指标 ,定量测定菜豆 (PhaseolusvulgarisL .)根构型在低磷胁迫下的适应性变化及其与磷效率的关系。结果表明 ,菜豆根构型对低磷胁迫具有适应性反应 ,在缺磷条件下基根向地性减弱 ,基根在生长介质表层相对分布增多、基根平均生长角度 (与水平线夹角 )变小 ,从而导致整个根系较浅。供试菜豆根构型对低磷胁迫的适应性反应具有显著基因型差异 ,缺磷时G19833等基因型向地性明显减弱 ,基根向高磷剖面趋向生长的能力较强 ,因而具有较高的磷吸收效率。研究结果表明根构型变化是菜豆适应低磷胁迫的可能机理之一 ,为通过改变植物根构型来提高磷吸收效率提供了依据。
Association mapping in structured populations
DOI:10.1086/302959
URL
PMID:10827107
[本文引用: 1]
The use, in association studies, of the forthcoming dense genomewide collection of single-nucleotide polymorphisms (SNPs) has been heralded as a potential breakthrough in the study of the genetic basis of common complex disorders. A serious problem with association mapping is that population structure can lead to spurious associations between a candidate marker and a phenotype. One common solution has been to abandon case-control studies in favor of family-based tests of association, such as the transmission/disequilibrium test (TDT), but this comes at a considerable cost in the need to collect DNA from close relatives of affected individuals. In this article we describe a novel, statistically valid, method for case-control association studies in structured populations. Our method uses a set of unlinked genetic markers to infer details of population structure, and to estimate the ancestry of sampled individuals, before using this information to test for associations within subpopulations. It provides power comparable with the TDT in many settings and may substantially outperform it if there are conflicting associations in different subpopulations.
An Arabidopsis example of association mapping in structured samples
DOI:10.1371/journal.pgen.0030004
URL
PMID:1779303
[本文引用: 1]
A potentially serious disadvantage of association mapping is the fact that marker-trait associations may arise from confounding population structure as well as from linkage to causative polymorphisms. Using genome-wide marker data, we have previously demonstrated that the problem can be severe in a global sample of 95Arabidopsis thalianaaccessions, and that established methods for controlling for population structure are generally insufficient. Here, we use the same sample together with a number of flowering-related phenotypes and data-perturbation simulations to evaluate a wider range of methods for controlling for population structure. We find that, in terms of reducing the false-positive rate while maintaining statistical power, a recently introduced mixed-model approach that takes genome-wide differences in relatedness into account via estimated pairwise kinship coefficients generally performs best. By combining the association results with results from linkage mapping in F2 crosses, we identify one previously known true positive and several promising new associations, but also demonstrate the existence of both false positives and false negatives. Our results illustrate the potential of genome-wide association scans as a tool for dissecting the genetics of natural variation, while at the same time highlighting the pitfalls. The importance of study design is clear; our study is severely under-powered both in terms of sample size and marker density. Our results also provide a striking demonstration of confounding by population structure. While statistical methods can be used to ameliorate this problem, they cannot always be effective and are certainly not a substitute for independent evidence, such as that obtained via crosses or transgenic experiments. Ultimately, association mapping is a powerful tool for identifying a list of candidates that is short enough to permit further genetic study. There is currently tremendous interest in using association mapping to find the genes responsible for natural variation, particularly for human disease. In association mapping, researchers seek to identify regions of the genome where individuals who are phenotypically similar (e.g., they all have the same disease) are also unusually closely related. A potentially serious problem is that spurious correlations may arise if the population is structured so that members of a subgroup tend to be much more closely related. We have previously demonstrated that this problem can be severe inArabidopsis thaliana,and that established statistical methods for controlling for population structure are insufficient. Here, we evaluate a broader range of methods. We find that a recently introduced mixed-model approach generally performs best. By combining the association results with results from linkage mapping in F2 crosses, we identify one previously known true positive and several promising new associations, but also demonstrate the existence of both false positives and false negatives. Our results illustrate the potential of genome-wide association scans as a tool for dissecting the genetics of natural variation, while at the same time highlighting the pitfalls.
Biochemical evidence bearing on the domestication of Phaseolus (Fabaceae) beans
DOI:10.1007/BF02860473
URL
[本文引用: 1]
The genus Phaseolus (Fabaceae) consists of some 50 species, all of which are distributed in the Americas. Four of these contain cultigens. P. vulgaris (common bean), P. lunatus (lima bean), P. acutifolius (tepary bean), P. coccineus subsp. coccineus (runner bean); and P. coccineus subsp. polyanthus (no English vernacular name). Biochemical markers - phaseolin seed storage protein and isozymes - have provided new evidence on the organization of the first three species. Domestication has possibly caused a strong reduction in genetic diversity in P. vulgaris and P. acutifolius. Both P. vulgaris and P. lunatus cultivars result from at least two independent domestications, in Mesoamerica and in the Andes. These two species consist of two gene pools, each of which includes wild ancestors and their respective cultivated descendants. Our findings suggest the need for additional emphasis on genetic conservation of wild ancestors and their use in breeding programs and for a comparison of inter-gene pool vs. intra-gene pool crosses in breeding programs. /// El género Phaseolus consta de unas 50 especies, distribuidas exclusivamente en las Américas; cuatro de estas especies incluyen a formas cultivadas: P. vulgaris (frijol común), P. lunatus (frijol lima), P. acutifolius (frijol tepari o escomite), P. coccineus subsp. coccineus (frijol ayocote), y P. coccineus subsp. polyanthus (frijol acalete). El uso de marcadores bioquímicos - faseolina e isozimas - ha producido nueva información acerca de la distribución de la variabilidad genética en las tres primeras especies. El proceso de domesticación causó una reducción marcada en la variabilidad genética para faseolina en P. vulgaris y P. acutifolius. Las formas cultivadas de P. vulgaris y P. lunatus resultaron de por lo menos dos domesticaciones distintas, en Mesoamérica y en los Andes. Estas dos especies constan de dos grupos de genotipos, cada cual incluye tanto a las formas silvestres ancestrales como a sus progenies cultivadas respectivas. Nuestros resultados sugieren que se ponga más enfasis tanto en la recolección y la preservación del germoplasma silvestre ancestral como en su uso en programas de mejoramiento; también sugieren que se compare en forma más detenida las cruzas entre grupos de genotipos con las cruzas adentro de estos grupos.
Genetic diversity of the common bacterial blight pathogen of bean,Xanthomonas axonopodis pv. phaseoli,in Iran revealed by rep-PCR and PCR-RFLP analyses
DOI:10.1007/s11274-011-0705-7
URL
[本文引用: 1]
Xanthomonad-like bacteria that are associated with common bacterial blight of bean in Iran were identified on the basis of their colonial morphology, biochemical and serological properties, presence of a specific DNA fragment using PCR primers and pathogenicity on bean. Xanthomonas axonopodis pv. phaseoli (Xap) strains were further characterized using rep-PCR and restriction fragment length polymorphism (RFLP). RFLP profiles generated by the restriction endonucleases RsaI, TaqI, HaeIII and Sau96I and rep-PCR analysis revealed that Iranian strains were relatively genetically homogenous. The similarity coefficients among the strains ranged from 0.87 to 1. The genetic diversity coefficients among strains from three infected provinces, Isfahan, Markazi and Lorestan, were 0.019, 0.072 and 0.033, respectively. The low overall level of polymorphism within Xap isolates collected from the three Iranian infected regions could suggest that few initial inoculum introductions might have distributed among these different bean-growing areas in Iran.
QTL mapping of root hair and acid exudation traits and their relationship to phosphorus uptake in common bean
DOI:10.1007/s11104-005-0693-1 URL [本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}