


开放科学(资源服务)标识码(OSID): 
WRKY转录因子是植物特有的最大的转录因子家族之一,属于DNA结合蛋白。WRKY转录因子最早发现于甘薯的SPF1[1],其后相继在拟南芥[2,3]、水稻[4,5]、小麦[6]和大麦[7]、玉米[8]、大豆[9]、马铃薯[10]、谷子[11]、甜菜[12]和棉花[13]等模式植物和多种作物上得到鉴定。在植物不同发育时期和多种环境因素诱导下,WRKY转录因子可通过正负调控蛋白调控植物生长发育、生物与非生物胁迫应答等[14,15]。WRKY转录因子在增强植物抗旱性方面扮演着重要角色,直接或间接参与植物抗旱应答调控。研究[3,16-20]发现,单独或协同调控拟南芥AtWRKY18、AtWRKY40、AtWRKY57和AtWRKY60等基因的表达,可增强拟南芥的耐旱能力;超表达OsWRKY11、OsWRKY30、OsWRKY45和OsWRKY47等基因可提高水稻的耐旱性[21,22,23,24,25];在小麦中,超表达Tawrky2可增强转基因植株的抗旱性[26];在玉米中也发现,过表达在水分胁迫响应中扮演重要角色的WRKY家族成员Zmwrky106基因有助于玉米耐旱能力的提高[27];以大豆中克隆到的WRKY家族成员Gmwrky35基因转化烟草,证实Gmwrky35具有增强抗旱能力作用[28]。
甜菜(Beta vulgaris L.)是我国重要的糖料作物和经济作物之一。甜菜主要种植在我国东北、西北和华北等区域,而周期性干旱天气频发、水资源相对匮乏等是这些地区的主要特点,干旱是制约甜菜产量和质量提升及产业可持续发展的主要因素之一[29]。目前,已经对甜菜WRKY转录因子全基因组进行了鉴定,发现有40个成员在9条染色体上呈不均匀分布,初步推测可能有5个基因参与盐胁迫调控,7个基因参与热胁迫调控[12];而有关甜菜WRKY转录因子家族基因与抗旱相关的研究还鲜见报道。本课题组在对甜菜水分胁迫下转录组分析中发现,甜菜WRKY转录因子家族成员BvWRKY23基因显著上调,推测其可能参与甜菜的抗旱应答,从甜菜中成功克隆了该基因,并构建了表达载体。鉴于上述前期工作,本研究构建以甜菜BvWRKY23为靶目标、含有反向重复发卡结构的RNAi表达载体,为进一步鉴定BvWRKY23基因在甜菜抗旱中的功能及其作用机制奠定基础。
1 材料与方法
1.1 试验材料
试验材料为内蒙古农业大学甜菜生理研究所筛选的甜菜抗旱材料HI0466,品种抗旱性鉴定工作已先期开展[30]。选取籽粒饱满的甜菜种子,用紫外灯照射2h后在育苗盘内播种,置于人工气候室培养,16h光照,8h暗期,温度25℃。在幼苗生长至6片真叶时进行胁迫处理,其中对照(CK)保持正常供水,胁迫处理停止浇水,分别于处理后4(DS4)、8(DS8)、10d(DS10)和复水后2d(RW2)取对照和处理的甜菜叶片,于-70℃保存用于转录组测序和RNA提取。
1.2 主要试剂
主要试剂有RNA提取试剂盒RNAplant plus reagent、Taq DNA聚合酶、限制性内切酶、T4 DNA连接酶和AMV反转录试剂盒、DNA凝胶回收和质粒DNA提取试剂盒、pUCm-T载体、PBSK-RTM中间载体、植物表达载体pCambia2301ky、氨苄青霉素(Ampicillin,AMP)、卡那霉素(Kanamycin)和大肠杆菌DH5α感受态细胞等。
1.3 引物设计及干扰片段的扩增
根据已克隆到的BvWRKY23基因全长选取RNAi靶片段,靶片段起始于起始密码子后10bp处,止于起始密码子后389bp处,共380bp。根据所选靶片段设计特异性正向引物P1和反向引物P2,然后分别在引物的5′端添加酶切位点BamHⅠ/XbaⅠ和SacⅠ/NotⅠ(下划线部分),并增加保护碱基(表1),保证正向片段和反向片段正确连接到中间载体PBSK-RTM。
表1 RNAi片段及内含子扩增引物
Table 1
| 引物类型Primer type | 引物名称Primer name | 序列(5'-3') Sequence (5'-3') | 酶切位点Enzyme site |
|---|---|---|---|
| P1 | BvWRKY23-F1 | CGGGATCCGAACCATTGAAGATTGAA | BamHI |
| BvWRKY23-R1 | GCTCTAGAAGAGGTGGAGGGGTAGGA | XbaI | |
| P2 | BvWRKY23-F2 | CGAGCTCGAACCATTGAAGATTGAA | SacI |
| BvWRKY23-R2 | ATTTGCGGCCGCAGAGGTGGAGGGGTAGGA | NotI | |
| 内含子Intron (RTM) | RTM-F | ACGTTGTAAGTCTATTTTTG | |
| RTM-R | TCTATCTGCTGGGTCCAAATC |
提取甜菜幼苗叶片总RNA,反转录合成cDNA,分别用引物P1和P2进行干扰靶片段的扩增。PCR反应程序为:98℃预变性2min;98℃变性10s,55℃退火15s,68℃延伸48s,35个循环;68℃延伸10min,4℃保存。PCR产物经1.5%琼脂糖凝胶电泳检测。通过DNA凝胶回收试剂盒回收目的片段,并将其连接到pUCm-T载体,用连接产物转化大肠杆菌DH5α感受态细胞,在100μg/mL AMP的LB平板上涂布IPTG和X-gal,于37℃倒置培养过夜,进行蓝白斑筛选,挑选白色菌落培养并进行PCR鉴定,将鉴定后的阳性克隆进行测序。引物合成和测序工作委托北京六合华大基因科技有限公司完成。
1.4 BvWRKY23-RNAi表达载体的构建
连接至pUCm-T载体上的干扰片段经测序确认正确后,用限制性内切酶BamHⅠ/XbaⅠ从pUCm-T载体上切下正向片段并将其连接到中间载体PBSK-RTM,构建载体PBSK-RTM-F。转化大肠杆菌DH5α,挑取单菌落,提取质粒并进行菌液PCR验证后测序。用限制性内切酶SacⅠ/NotⅠ从pUCm-T载体上切下反向片段,并将其连接至载体PBSK-RTM-F对应的多克隆位点之间,构建中间载体PBSK-RTM-FR,转化大肠杆菌DH5α,挑取单菌落,提取质粒并进行菌液PCR验证及测序。将干扰片段以相反的方向连接到中间载体内含子两侧之后,用限制性内切酶BamHⅠ和SacⅠ酶切已构建好的中间载体PBSK-RTM-FR和干扰表达载体pCambia2301ky。将中间载体PBSK-RTM-FR上的正向干扰片段-内含子(RTM)-反向干扰片段连接至表达载体pCambia2301ky对应酶切位点之间,构建干扰表达载体pCambia2301ky-BvWRKY23-RNAi(图1),转化大肠杆菌DH5α,随机挑取8个转化子,通过正向片段和内含子引物进行菌液PCR验证,挑取阳性的转化子用于质粒提取,通过BamHⅠ和SacⅠ进行双酶切鉴定。
图1
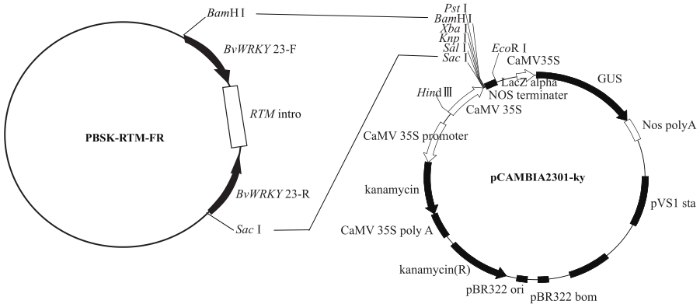
2 结果与分析
2.1 水分胁迫下BvWRKY23基因的表达情况
通过转录组测序(RNA-seq)技术对不同水分处理条件下甜菜幼苗叶片BvWRKY23基因进行表达定量分析,从以FPKM(fragments per kilobase per million,每千个碱基的转录每百万映射读取的片段数)为单位的BvWRKY23基因表达定量结果(图2)表明,随着水分胁迫程度的加剧,BvWRKY23基因的表达量也逐步增强,在胁迫程度最严重的第10天表达量最高,为CK表达量的5.3倍。而复水后随着胁迫信号的解除,该基因的表达量显著降低,表明BvWRKY23基因对水分胁迫高度敏感,水分胁迫特异性诱导该基因的过量表达,由此推测该基因可能具有提高甜菜抗旱能力的作用。
图2
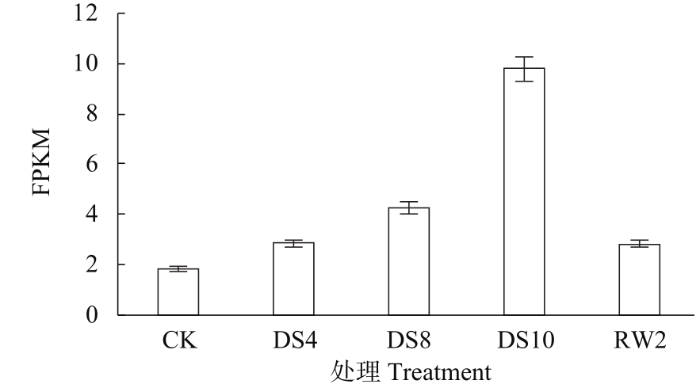
图2
不同水分胁迫对甜菜叶片BvWRKY23基因表达的影响
Fig.2
Effects of water stress on BvWRKY23 gene expression in sugar beet leaves
2.2 BvWRKY23基因干扰片段的克隆
以甜菜叶片总RNA反转录所获得的cDNA为模板,通过正、反向引物进行PCR扩增,最终获得2条约380bp的干扰片段,与所选取干扰片段的大小相同(图3)。将扩增片段分别进行胶回收、纯化,并连接到pUCm-T载体上进行测序,通过GeneBank的BLAST(http//blast.ncbi.nlm.nih.gov/Blast.cgi)功能将测序结果与BvWRKY23的基因序列进行比对,结果显示比对结果完全吻合,表明已成功克隆到干扰片段。
图3
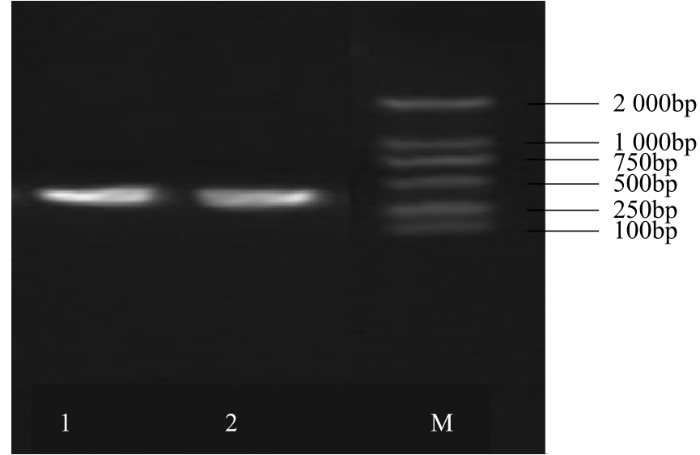
图3
PCR产物BvWRKY23基因的RNAi片段
M:DL2000,下同;1~2:RNAi片段产物
Fig.3
PCR products of BvWRKY23 gene RNAi fragments
M: DL2000 Marker, the same below; 1-2: The products of RNAi fragments
2.3 中间载体的构建
图4
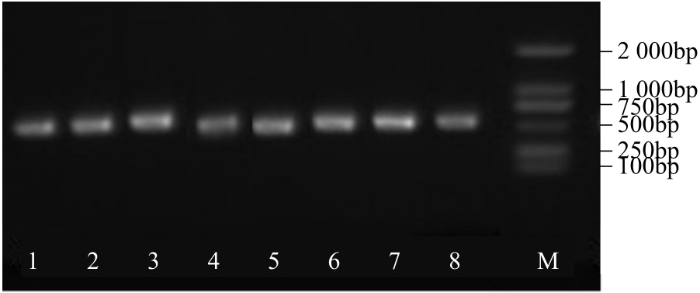
图4
中间载体PBSK-RTM-F阳性克隆菌落的PCR鉴定
1~8:PCR扩增干扰片段。下同
Fig.4
The PCR identification of positive clones for intermediate vector PBSK-RTM-F
1-8: The PCR products of interference fragment. The same below
图5

图5
PBSK-RTM-F阳性质粒测序
第一行为参考序列;第二行为测序序列。下同
Fig.5
Sequencing analyses of positive clones PBSK-RTM-F
The first line is the reference sequence, the second line is the sequencing. The same below
图6
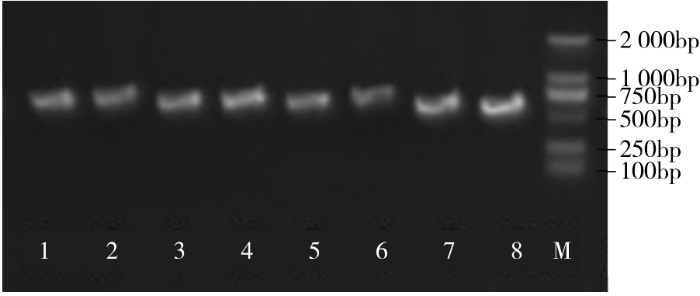
图6
中间载体PBSK-RTM-FR阳性克隆菌落PCR 鉴定
Fig.6
The PCR identification of positive clones for intermediate vector PBSK-RTM-FR
图7
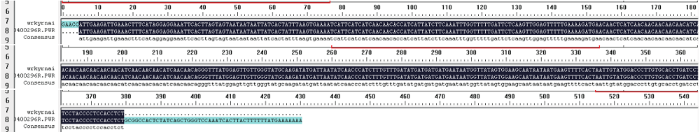
图7
中间载体PBSK-RTM-FR阳性质粒测序
Fig.7
Sequencing analysis of positive clones of intermediate vector PBSK-RTM-FR
2.4 BvWRKY23 RNAi载体的构建及酶切鉴定
BamHⅠ/SacⅠ双酶切PBSK-RTM-FR中间载体,回收大片段(正向片段+RTM+反向片段)。同时BamHⅠ/SacⅠ双酶切pCambia2301ky质粒,将回收的大片段与表达载体连接后构建pCambia2301ky-BvWRKY23-RNAi载体,并转化大肠杆菌DH5α,然后随机挑取8个转化子,通过正向片段和内含子(RTM)引物进行菌液PCR验证,获得6个阳性转化子,大小为508bp,正好是正向片段与内含子之和(图8)。
图8
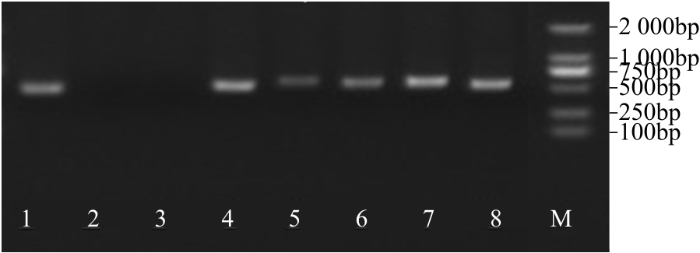
图8
pCambia2301ky-BvWRKY23-RNAi载体阳性重组PCR鉴定
1~8:不同转化子PCR鉴定
Fig.8
The identification of positive recombinant clone for pCambia2301ky-BvWRKY23-RNAi by PCR
1-8: The PCR identification of different transformants
挑取1个PCR阳性的转化子进行质粒提取,通过BamHⅠ/SacⅠ双酶切pCambia2301ky-BvWRKY23-RNAi质粒进行鉴定,电泳结果显示,出现质粒骨架条带和干扰片段条带。其中在750~1 000bp的条带大小为888bp,与正向干扰片段+RTM(内含子)+反向干扰片段大小相符,而双酶切空载体仅出现质粒骨架条带(图9),表明从中间载体PBSK-RTM-FR上切下的正向干扰片段+内含子(RTM)+反向干扰片段已经成功连接到表达载体pCambia2301ky质粒骨架上,即RNAi载体构建完成。
图9
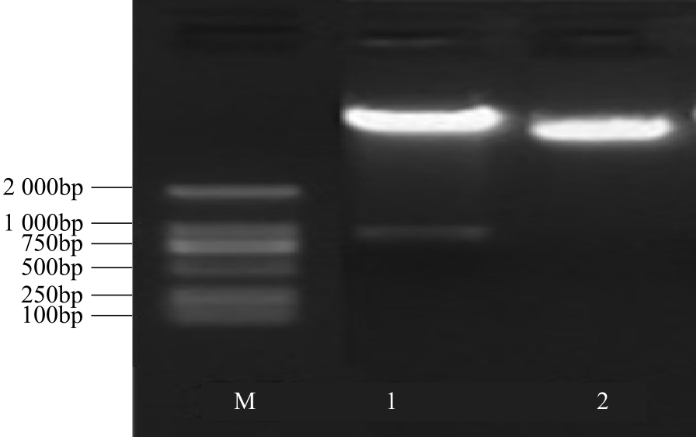
图9
pCambia2301ky-BvWRKY23-RNAi载体BamHⅠ/SacⅠ双酶切鉴定
1:BamHⅠ/SacⅠ双酶切pCambia2301ky-BvWRKY23-RNAi;2:BamHⅠ/SacⅠ双酶切空载质粒
Fig.9
The identification of plasmid pCambia2301ky-BvWRKY23-RNAi vector by double digestion with BamHⅠ/SacⅠ
1: Double enzyme digestion products of pCambia2301ky-BvWRKY23-RNAi by BamHⅠ/SacⅠ; 2: Double enzyme digestion products of pCambia2301ky by BamHⅠ/SacⅠ
3 讨论
在利用RNAi技术开展相关研究过程中,构建相应的RNAi表达载体至关重要。构建植物RNAi载体的方法包括传统的“酶切-连接”法[43]、“零背景筛选”技术[44]、Gateway技术[45]和人工miRNA技术[46]。最早的构建RNA干扰载体的方法是传统的“酶切-连接”法,已在多种植物中成功构建了RNA干扰表达载体[47,48,49,50],但该技术要求原始载体至少有4个可用酶切位点,且仅可在载体上插入1~2个基因,同时对目的片段的限制条件较多,增加了原始载体和目的片段的选择难度;“零背景筛选”技术可大大降低对原载体可用酶切位点的要求,通过简单的2步PCR反应就能形成干扰载体所需的“发夹”结构,但该技术对引物设计和DNA聚合酶有较高的要求;Gateway技术虽然可以快速、高效地构建植物RNAi表达载体,但高额的专用酶成本在一定程度上限制了其应用[51]。人工miRNA技术具有高效、精确和可控等优点,被认为是替代RNAi的有效工具,但由于某些特定植物基因组信息的缺乏,沉默效应还不能完全做到人为控制。因此,在构建RNAi表达载体时,应根据试验具体情况和要求选择适宜的载体构建方法。
4 结论
WRKY转录因子家族成员在调控植物生长发育、应答生物与非生物胁迫等方面具有重要的生物学功能。通过传统的“酶切-连接”方法成功构建了甜菜WRKY转录因子家族成员BvWRKY23基因的RNAi表达载体。
参考文献
Characterization of a cDNA encoding a novel DNA-binding protein,SPF1,that recognizes SP8 sequences in the 5’ upstream regions of genes coding for sporamin and beta-amylase from sweet potato
Expression profile matrix of Arabidopsis transcription factor genes suggests their putative functions in response to environmental stresses
DOI:10.1105/tpc.010410
URL
PMID:11910004
[本文引用: 1]

Numerous studies have shown that transcription factors are important in regulating plant responses to environmental stress. However, specific functions for most of the genes encoding transcription factors are unclear. In this study, we used mRNA profiles generated from microarray experiments to deduce the functions of genes encoding known and putative Arabidopsis transcription factors. The mRNA levels of 402 distinct transcription factor genes were examined at different developmental stages and under various stress conditions. Transcription factors potentially controlling downstream gene expression in stress signal transduction pathways were identified by observed activation and repression of the genes after certain stress treatments. The mRNA levels of a number of previously characterized transcription factor genes were changed significantly in connection with other regulatory pathways, suggesting their multifunctional nature. The expression of 74 transcription factor genes responsive to bacterial pathogen infection was reduced or abolished in mutants that have defects in salicylic acid, jasmonic acid, or ethylene signaling. This observation indicates that the regulation of these genes is mediated at least partly by these plant hormones and suggests that the transcription factor genes are involved in the regulation of additional downstream responses mediated by these hormones. Among the 43 transcription factor genes that are induced during senescence, 28 of them also are induced by stress treatment, suggesting extensive overlap responses to these stresses. Statistical analysis of the promoter regions of the genes responsive to cold stress indicated unambiguous enrichment of known conserved transcription factor binding sites for the responses. A highly conserved novel promoter motif was identified in genes responding to a broad set of pathogen infection treatments. This observation strongly suggests that the corresponding transcription factors play general and crucial roles in the coordinated regulation of these specific regulons. Although further validation is needed, these correlative results provide a vast amount of information that can guide hypothesis-driven research to elucidate the molecular mechanisms involved in transcriptional regulation and signaling networks in plants.
Physical and functional interactions between pathogen-induced Arabidopsis WRKY18,WRKY40,and WRKY60 transcription factors
DOI:10.1105/tpc.105.037523
URL
PMID:16603654
[本文引用: 2]
Limited information is available about the roles of specific WRKY transcription factors in plant defense. We report physical and functional interactions between structurally related and pathogen-induced WRKY18, WRKY40, and WRKY60 transcription factors in Arabidopsis thaliana. The three WRKY proteins formed both homocomplexes and heterocomplexes and DNA binding activities were significantly shifted depending on which WRKY proteins were present in these complexes. Single WRKY mutants exhibited no or small alterations in response to the hemibiotrophic bacterial pathogen Pseudomonas syringae and the necrotrophic fungal pathogen Botrytis cinerea. However, wrky18 wrky40 and wrky18 wrky60 double mutants and the wrky18 wrky40 wrky60 triple mutant were substantially more resistant to P. syringae but more susceptible to B. cinerea than wild-type plants. Thus, the three WRKY proteins have partially redundant roles in plant responses to the two distinct types of pathogens, with WRKY18 playing a more important role than the other two. The contrasting responses of these WRKY mutants to the two pathogens correlated with opposite effects on pathogen-induced expression of salicylic acid-regulated PATHOGENESIS-RELATED1 and jasmonic acid-regulated PDF1.2. While constitutive expression of WRKY18 enhanced resistance to P. syringae, its coexpression with WRKY40 or WRKY60 made plants more susceptible to both P. syringae and B. cinerea. These results indicate that the three WRKY proteins interact both physically and functionally in a complex pattern of overlapping, antagonistic, and distinct roles in plant responses to different types of microbial pathogens.
A comprehensive transcriptional profiling of the WRKY gene family in rice under various abiotic and phytohormone treatments
DOI:10.1093/pcp/pcn061
URL
PMID:18413358
[本文引用: 1]
WRKY transcription factors play important roles in the regulation of various biological processes. We have analyzed the publicly available rice genome sequence databases and predicted 103 genes encoding WRKY transcription factors. Among them, the majority of rice WRKY genes (77.7%) were located in duplicated regions; 45.6% of WRKY genes were fragmentally duplicated and 35% of them were tandemly duplicated. These results suggested that genome duplications might be regarded as a major mechanism for expansion of this family in the rice genome. Subsequently, we analyzed their expression profiles under normal and abiotic stress, as well as various hormone treatments. Under normal growth conditions, 65 WRKY genes were expressed differentially either in their transcript abundance or in their expression patterns. Under abiotic (cold, drought and salinity) stresses and various phytohormone treatments, 54 WRKY genes exhibited significant differences in their transcript abundance; among them three genes were expressed only in stressed conditions. Among the stress-inducible genes, 13 genes were regulated only by abiotic stresses, another set of 13 genes were responsive to only phytohormone treatments and the remaining 28 genes were regulated by both factors, suggesting an interaction between abiotic stress and hormone signaling. On the other hand, we have also surveyed the expression divergence of duplicated genes under normal or stressed conditions, and the results showed that high expression divergence has occurred not only among fragmentally but also among tandemly duplicated genes. These results suggested that the high expression divergence could be one of the mechanisms for the retention of these duplicated WRKY genes.
The WRKY gene family in rice (Oryza sativa)
DOI:10.1111/j.1744-7909.2007.00504.x
URL
[本文引用: 1]
WRKY genes encode transcription factors that are involved in the regulation of various biological processes. These zinc-finger proteins, especially those members mediating stress responses, are uniquely expanded in plants. To facilitate the study of the evolutionary history and functions of this supergene family, we performed an exhaustive search for WRKY genes using HMMER and a Hidden Markov Model that was specifically trained for rice. This work resulted in a comprehensive list of WRKY gene models in Oryza sativa L. ssp. indica and L. ssp. japonica. Mapping of these genes to individual chromosomes facilitated elimination of the redundant, leading to the identification of 98 WRKY genes in japonica and 102 in indica rice. These genes were further categorized according to the number and structure of their zinc-finger domains. Based on a phylogenetic tree of the conserved WRKY domains and the graphic display of WRKY loci on corresponding indica and japonica chromosomes, we identified possible WRKY gene duplications within, and losses between the two closely related rice subspecies. Also reviewed are the roles of WRKY genes in disease resistance and responses to salicylic acid and jasmonic acid, seed development and germination mediated by gibberellins, other developmental processes including senescence, and responses to abiotic stresses and abscisic acid in rice and other plants. The signaling pathways mediating WRKY gene expression are also discussed.
Genome-wide analysis of WRKY transcription factors in wheat (Triticum aestivum L.) and differential expression under water deficit condition
DOI:10.7717/peerj.3232
URL
PMID:28484671
[本文引用: 1]
BACKGROUND: WRKY proteins, which comprise one of the largest transcription factor (TF) families in the plant kingdom, play crucial roles in plant development and stress responses. Despite several studies on WRKYs in wheat (Triticum aestivum L.), functional annotation information about wheat WRKYs is limited. RESULTS: Here, 171 TaWRKY TFs were identified from the whole wheat genome and compared with proteins from 19 other species representing nine major plant lineages. A phylogenetic analysis, coupled with gene structure analysis and motif determination, divided these TaWRKYs into seven subgroups (Group I, IIa-e, and III). Chromosomal location showed that most TaWRKY genes were enriched on four chromosomes, especially on chromosome 3B. In addition, 85 (49.7%) genes were either tandem (5) or segmental duplication (80), which suggested that though tandem duplication has contributed to the expansion of TaWRKY family, segmental duplication probably played a more pivotal role. Analysis of cis-acting elements revealed putative functions of WRKYs in wheat during development as well as under numerous biotic and abiotic stresses. Finally, the expression of TaWRKY genes in flag leaves, glumes, and lemmas under water-deficit condition were analyzed. Results showed that different TaWRKY genes preferentially express in specific tissue during the grain-filling stage. CONCLUSION: Our results provide a more extensive insight on WRKY gene family in wheat, and also contribute to the screening of more candidate genes for further investigation on function characterization of WRKYs under various stresses.
Phylogenetic and comparative gene expression analysis of barley (Hordeum vulgare) WRKY transcription factor family reveals putatively retained functions between monocots and dicots
DOI:10.1186/1471-2164-9-194 URL [本文引用: 1]
Molecular phylogenetic and expression analysis of the complete WRKY transcription factor family in maize
DOI:10.1093/dnares/dsr048
URL
[本文引用: 1]
15% are further analysed to discover new organ- or tissue-specific genes. In addition, microarray analyses of transcriptional responses to drought stress and fungal infection showed that maize WRKY proteins are involved in stress responses. All these results contribute to a deep probing into the roles of WRKY transcription factors in maize growth and development and stress tolerance.]]>
Characterization of soybean WRKY gene family and identification of soybean WRKY genes that promote resistance to soybean cyst nematode
DOI:10.1038/s41598-017-18235-8
URL
PMID:29259331
[本文引用: 1]
WRKY proteins are a superfamily of plant transcription factors with important roles in plants. WRKY proteins have been extensively analyzed in plant species including Arabidopsis and rice. Here we report characterization of soybean WRKY gene family and their functional analysis in resistance to soybean cyst nematode (SCN), the most important soybean pathogen. Through search of the soybean genome, we identified 174 genes encoding WRKY proteins that can be classified into seven groups as established in other plants. WRKY variants including a WRKY-related protein unique to legumes have also been identified. Expression analysis reveals both diverse expression patterns in different soybean tissues and preferential expression of specific WRKY groups in certain tissues. Furthermore, a large number of soybean WRKY genes were responsive to salicylic acid. To identify soybean WRKY genes that promote soybean resistance to SCN, we first screened soybean WRKY genes for enhancing SCN resistance when over-expressed in transgenic soybean hairy roots. To confirm the results, we transformed five WRKY genes into a SCN-susceptible soybean cultivar and generated transgenic soybean lines. Transgenic soybean lines overexpressing three WRKY transgenes displayed increased resistance to SCN. Thus, WRKY genes could be explored to develop new soybean cultivars with enhanced resistance to SCN.
A potato gene,erg-1,is rapidly induced by Erwinia carotovora ssp. atroseptica,Phytophthora infestans,ethylene and salicylic acid
DOI:10.1016/S0176-1617(00)80191-1 URL [本文引用: 1]
谷子WRKY36转录因子的分子特性及功能鉴定
DOI:10.3864/j.issn.0578-1752.2015.05.03
URL
[本文引用: 1]
SiWRKY36的分子特性和功能,解析谷子转录因子的抗逆调控机制。【方法】通过对干旱胁迫谷子转录组测序结果分析,获得了一个WRKY转录因子SiWRKY36;利用生物信息学的方法分析谷子SiWRKY36的分子特性;根据SiWRKY36蛋白序列进行同源性搜索,得到与谷子SiWRKY36蛋白序列相似度较高的其他物种的蛋白序列;使用MEGA5对谷子SiWRKY36蛋白序列及其同源序列进行多序列比对分析并构建同源物种间系统进化树;利用MEME和SMART在线工具进行蛋白序列分析;利用GSDS和PHYRE2在线工具分别对谷子SiWRKY36基因结构和三级结构进行分析;从谷子基因组数据库Phytozome获取谷子SiWRKY36上游2 000 bp作为启动子;用PLACE数据库对SiWRKY36启动子顺式作用元件进行分析;利用实时荧光定量PCR检测SiWRKY36在不同胁迫条件下(PEG、低温、NaCl、MeJA、ABA、GA和SA、H2O2)的表达模式;分别以8种胁迫处理的谷子cDNA作为模板,以谷子Si001873m.g为内参,以SYBR Green染料法进行real-time PCR。用实时荧光定量PCR仪进行PCR扩增;将SiWRKY36的cDNA序列连入带有CaMV 35S启动子的pBI121表达载体中,构建表达载体pBI121-SiWRKY36,转入农杆菌,侵染野生型拟南芥得到转基因株系。用T3转SiWRKY36拟南芥植株进行抗性鉴定。【结果】谷子SiWRKY36全长1 485 bp,基因编码区包含UTR区和3个内含子以及4个外显子,与柳枝稷亲缘性最高,属于WRKY转录因子家族的第一类。SiWRKY36编码蛋白包含2个WRKY保守域,预测的SiWRKY36蛋白三级结构包含2个α螺旋结构和3个β折叠结构。启动子元件分析表明SiWRKY36包含ABA-responsive element(ABRE)、MYB、MYC、low-temperature-responsive element(LTRE)、GT-1等多种逆境胁迫应答元件。实时荧光定量PCR结果显示SiWRKY36对多种非生物胁迫和激素均有不同程度的响应,但在H2O2和低温处理下基因表达量无明显变化;亚细胞定位结果表明SiWRKY36蛋白主要定位于细胞核中。抗性鉴定结果显示,在不进行任何处理的MS培养基上,野生型拟南芥和过表达株系拟南芥的长势基本一致;在2% PEG的处理条件下,3个转基因株系的根长、根的总表面积和根的总体积要大于野生型拟南芥。【结论】SiWRKY36转基因植株可能对轻度干旱有一定的抗性。]]>
甜菜WRKY转录因子全基因组鉴定及其在非生物胁迫下的表达分析
DOI:10.3864/j.issn.0578-1752.2017.17.002
URL
[本文引用: 2]
BvWRKYs),解析其组织特异性及盐、热胁迫下的表达情况,为该类基因的功能研究提供参考,为观赏甜菜和石竹目其他观赏植物的基因工程打下基础。【方法】以75条拟南芥WRKY蛋白为参考,根据WRKY保守蛋白序列(PF03106)利用hmm和BLAST同源性搜索对甜菜WRKY家族基因进行鉴定。利用MapInspect、GSDS2.0、MEGA5.0、DNAMAN5.0、WebLogo 3、MEME生物信息学工具对甜菜WRKY家族基因染色体定位、系统发生关系、基因结构、蛋白质保守结构域、保守元件进行预测和分析。利用RNA-seq和qRT-PCR分析甜菜WRKY组织表达特异性,盐胁迫、热胁迫条件下WRKY表达情况。【结果】甜菜WRKY家族基因包含40个成员,其中39条不均匀地分布在9条染色体上,另外1条定位到随机片段上。根据WRKY保守域特征并与拟南芥WRKY蛋白进化分析,可将40个成员分为Ⅰ、Ⅱ、Ⅲ 3类,Ⅰ类有9个成员,Ⅱ类有26个成员,Ⅲ类有5个成员。根据进化关系Ⅱ类可进一步分为Ⅱa(1个)、Ⅱb(4个)、Ⅱc(9个)、Ⅱd(5个)和Ⅱe(7个)5个亚类。基因结构分析发现,甜菜WRKY外显子和内含子数目具有高变异性(2—7个外显子),即使同一亚类内也都差异较大。保守元件分析显示同一类或亚类内成员具有相同的保守元件。WRKY保守域分析发现2个WRKY七肽域变型:WRKYGKK和WRKYGEK。每个WRKY至少在2个组织中表达,30个WRKY在叶中表达,40个WRKY在花序中均有表达,36个WRKY在幼叶中有表达,38个WRKY在直根中有表达,39个WRKY在幼苗中有表达,36个WRKY在种子中有表达。各WRKY表达量差异较大,可分为低表达、高表达基因两类,如BvWRKY23、BvWRKY3、BvWRKY11、BvWRKY7、BvWRKY6、BvWRKY26、BvWRKY4、BvWRKY40、BvWRKY24、BvWRKY2和BvWRKY28在各组织中均有较高表达,而BvWRKY38、BvWRKY13、BvWRKY36、BvWRKY35、BvWRKY5和BvWRKY34在各组织中均表达较低。热胁迫条件下BvWRKY16、BvWRKY21、BvWRKY20、BvWRKY22、BvWRKY32、BvWRKY33和BvWRKY34上调表达;盐胁迫条件下BvWRKY1、BvWRKY6、BvWRKY19、BvWRKY31和BvWRKY33呈现不同程度上调表达;BvWRKY33对热、盐2种胁迫均有明显响应。【结论】甜菜WRKY蛋白结构高度保守,基因序列长度和内含子数量变化很大,在不同组织中呈现出多种表达模式,部分WRKY响应热或盐胁迫,对甜菜逆境生理调控起重要作用。]]>
Transcriptome analysis reveals salt-stress-regulated biological processes and key pathways in roots of cotton (Gossypium hirsutum L.)
DOI:10.1016/j.ygeno.2011.04.007
URL
[本文引用: 1]
High salinity is one of the main factors limiting cotton growth and productivity. The genes that regulate salt stress in TM-1 upland cotton were monitored using microarray and real-time PCR (RT-PCR) with samples taken from roots. Microarray analysis showed that 1503 probe sets were up-regulated and 1490 probe sets were down-regulated in plants exposed for 3 h to 100 mM NaCl, and RT-PCR analysis validated 42 relevant/related genes. The distribution of enriched gene ontology terms showed such important processes as the response to water stress and pathways of hormone metabolism and signal transduction were induced by the NaCl treatment. Some key regulatory gene families involved in abiotic and biotic sources of stress such as WRKY, ERF, and JAZ were differentially expressed. Our transcriptome analysis might provide some useful insights into salt-mediated signal transduction pathways in cotton and offer a number of candidate genes as potential markers of tolerance to salt stress. (C) 2011 Elsevier Inc.
WRKY transcription factors:from DNA binding towards biological function
DOI:10.1016/j.pbi.2004.07.012
URL
[本文引用: 1]
WRKY genes seem to have originated in early eukaryotes. The cognate DNA-binding site of WRKY factors is well defined, but determining the roles of individual family members in regulating specific transcriptional programs during development or in response to environmental signals remains daunting. This review summarises the recent advances made in starting to unravel the various functions controlled by WRKY proteins.]]>
WRKY transcription factors
DOI:10.1016/j.tplants.2010.02.006
URL
[本文引用: 1]
WRKY transcription factors are one of the largest families of transcriptional regulators in plants and form integral parts of signalling webs that modulate many plant processes. Here, we review recent significant progress in WRKY transcription factor research. New findings illustrate that WRKY proteins often act as repressors as well as activators, and that members of the family play roles in both the repression and de-repression of important plant processes. Furthermore, it is becoming clear that a single WRKY transcription factor might be involved in regulating several seemingly disparate processes. Mechanisms of signalling and transcriptional regulation are being dissected, uncovering WRKY protein functions via interactions with a diverse array of protein partners, including MAP kinases, MAP kinase kinases, 14-3-3 proteins, calmodulin, histone deacetylases, resistance proteins and other WRKY transcription factors. WRKY genes exhibit extensive autoregulation and cross-regulation that facilitates transcriptional reprogramming in a dynamic web with built-in redundancy.
Activated expression of WRKY57 confers drought tolerance in Arabidopsis
DOI:10.1093/mp/sss080
URL
[本文引用: 1]
Drought is one of the most serious environmental factors that limit the productivity of agricultural crops worldwide. However, the mechanism underlying drought tolerance in plants is unclear. WRKY transcription factors are known to function in adaptation to abiotic stresses. By screening a pool of WRKY-associated T-DNA insertion mutants, we isolated a gain-of-function mutant, acquired drought tolerance (adt), showing improved drought tolerance. Under drought stress conditions, adt accumulated higher levels of ABA than wild-type plants. Stomatal aperture analysis indicated that adt was more sensitive to ABA than wild-type plants. Molecular genetic analysis revealed that a T-DNA insertion in adt led to activated expression of a WRKY gene that encodes the WRKR57 protein. Constitutive expression of WRKY57 also conferred similar drought tolerance. Consistently with the high ABA content and enhanced drought tolerance, three stress-responsive genes (RD29A, NCED3, and ABA3) were up-regulated in adt. ChIP assays demonstrated that WRKY57 can directly bind the W-box of RD29A and NCED3 promoter sequences. In addition, during ABA treatment, seed germination and early seedling growth of adt were inhibited, whereas, under high osmotic conditions, adt showed a higher seed germination frequency. In summary, our results suggested that the activated expression of WRKY57 improved drought tolerance of Arabidopsis by elevation of ABA levels. Establishment of the functions of WRKY57 will enable improvement of plant drought tolerance through gene manipulation approaches.
Roles of Arabidopsis WRKY18,WRKY40 and WRKY60 transcription factors in plant responses to abscisic acid and abiotic stress
DOI:10.1186/1471-2229-10-281
URL
PMID:21167067
BACKGROUND: WRKY transcription factors are involved in plant responses to both biotic and abiotic stresses. Arabidopsis WRKY18, WRKY40, and WRKY60 transcription factors interact both physically and functionally in plant defense responses. However, their role in plant abiotic stress response has not been directly analyzed. RESULTS: We report that the three WRKYs are involved in plant responses to abscisic acid (ABA) and abiotic stress. Through analysis of single, double, and triple mutants and overexpression lines for the WRKY genes, we have shown that WRKY18 and WRKY60 have a positive effect on plant ABA sensitivity for inhibition of seed germination and root growth. The same two WRKY genes also enhance plant sensitivity to salt and osmotic stress. WRKY40, on the other hand, antagonizes WRKY18 and WRKY60 in the effect on plant sensitivity to ABA and abiotic stress in germination and growth assays. Both WRKY18 and WRKY40 are rapidly induced by ABA, while induction of WRKY60 by ABA is delayed. ABA-inducible expression of WRKY60 is almost completely abolished in the wrky18 and wrky40 mutants. WRKY18 and WRKY40 recognize a cluster of W-box sequences in the WRKY60 promoter and activate WRKY60 expression in protoplasts. Thus, WRKY60 might be a direct target gene of WRKY18 and WRKY40 in ABA signaling. Using a stable transgenic reporter/effector system, we have shown that both WRKY18 and WRKY60 act as weak transcriptional activators while WRKY40 is a transcriptional repressor in plant cells. CONCLUSIONS: We propose that the three related WRKY transcription factors form a highly interacting regulatory network that modulates gene expression in both plant defense and stress responses by acting as either transcription activator or repressor.
Transcriptional reprogramming regulated by WRKY18 and WRKY40 facilitates powdery mildew infection of Arabidopsis
DOI:10.1111/j.1365-313X.2010.04387.x
URL
PMID:21143673
The two closely related Arabidopsis transcription factors, WRKY18 and WRKY40, play a major and partly redundant role in PAMP-triggered basal defense. We monitored the transcriptional reprogramming induced by the powdery mildew fungus, Golovinomyces orontii, during early stages of infection with respect to the role of WRKY18/40. Expression of >1300 Arabidopsis genes was differentially altered already 8 hours post infection (hpi), indicating rapid pre-penetration signaling between the pathogen and the host. We found that WRKY18/40 negatively affects pre-invasion host defenses and deduced a subset of genes that appear to be under WRKY18/40 control. A mutant lacking the WRKY18/40 repressors executes pathogen-dependent but exaggerated expression of some defense genes leading, for example, to strongly elevated levels of camalexin. This implies that WRKY18/40 act in a feedback repression system controlling basal defense. Moreover, using chromatin immunoprecipitation (ChIP), direct in vivo interactions of WRKY40 to promoter regions containing W box elements of the regulatory gene EDS1, the AP2-type transcription factor gene RRTF1 and to JAZ8, a member of the JA-signaling repressor gene family were demonstrated. Our data support a model in which WRKY18/40 negatively modulate the expression of positive regulators of defense such as CYP71A13, EDS1 and PAD4, but positively modulate the expression of some key JA-signaling genes by partly suppressing the expression of JAZ repressors.
Analyses of wrky18 wrky40 plants reveal critical roles of SA/EDS1 signaling and indole-glucosinolate biosynthesis for Golovinomyces orontii resistance and a loss-of resistance towards Pseudomonas syringae pv. tomato AvrRPS4
DOI:10.1094/MPMI-11-12-0265-R
URL
PMID:23617415
Simultaneous mutation of two WRKY-type transcription factors, WRKY18 and WRKY40, renders otherwise susceptible wild-type Arabidopsis plants resistant towards the biotrophic powdery mildew fungus Golovinomyces orontii. Resistance in wrky18 wrky40 double mutant plants is accompanied by massive transcriptional reprogramming, imbalance in salicylic acid (SA) and jasmonic acid (JA) signaling, altered ENHANCED DISEASE SUSCEPTIBILITY1 (EDS1) expression, and accumulation of the phytoalexin camalexin. Genetic analyses identified SA biosynthesis and EDS1 signaling as well as biosynthesis of the indole-glucosinolate 4MI3G as essential components required for loss-of-WRKY18 WRKY40-mediated resistance towards G. orontii. The analysis of wrky18 wrky40 pad3 mutant plants impaired in camalexin biosynthesis revealed an uncoupling of pre- from postinvasive resistance against G. orontii. Comprehensive infection studies demonstrated the specificity of wrky18 wrky40-mediated G. orontii resistance. Interestingly, WRKY18 and WRKY40 act as positive regulators in effector-triggered immunity, as the wrky18 wrky40 double mutant was found to be strongly susceptible towards the bacterial pathogen Pseudomonas syringae DC3000 expressing the effector AvrRPS4 but not against other tested Pseudomonas strains. We hypothesize that G. orontii depends on the function of WRKY18 and WRKY40 to successfully infect Arabidopsis wild-type plants while, in the interaction with P. syringae AvrRPS4, they are required to mediate effector-triggered immunity.
Co-expression of AtbHLH17 and AtWRKY28 confers resistance to abiotic stress in Arabidopsis
DOI:10.1007/s11248-012-9645-8
URL
[本文引用: 1]
Stress adaptation in plants involves altered expression of many genes through complex signaling pathways. To achieve the optimum expression of downstream functional genes, we expressed AtbHLH17 (AtAIB) and AtWRKY28 TFs which are known to be upregulated under drought and oxidative stress, respectively in Arabidopsis. Multigene expression cassette with these two TFs and reporter gene GUS was developed using modified gateway cloning strategy. The GUS assay and expression analysis of transgenes in transgenic plants confirmed the integration of multigene cassette. The transgenic lines exhibited enhanced tolerance to NaCl, Mannitol and oxidative stress. Under mannitol stress condition significantly higher root growth was observed in transgenics. Growth under stress and recovery growth was substantially superior in transgenics exposed to gradual long term desiccation stress conditions. We demonstrate the expression of several downstream target genes under various stress conditions. A few genes having either WRKY or bHLH cis elements in their promoter regions showed higher transcript levels than wild type. However, the genes which did not have either of the motifs did not differ in their expression levels in stress conditions compared to wild type plants. Hence co-expressing two or more TFs may result in upregulation of many downstream target genes and substantially improve the stress tolerance of the plants.]]>
Enhanced heat and drought tolerance in transgenic rice seedlings overexpressing OsWRKY11 under the control of HSP101 promoter
DOI:10.1007/s00299-008-0614-x
URL
[本文引用: 1]
An OsWRKY11 gene, which encodes a transcription factor with the WRKY domain, was identified as one of the genes that was induced by both heat shock and drought stresses in seedlings of rice (Oryza sativa L.). To determine if overexpression of OsWRKY11 confers heat and drought tolerance, OsWRKY11 cDNA was fused to the promoter of HSP101 of rice and introduced into a rice cultivar Sasanishiki. Overexpression of OsWRKY11 was induced by heat treatment. After heat pretreatment, the transgenic lines showed significant heat and drought tolerance, as indicated by the slower leaf-wilting and less-impaired survival rate of green parts of plants. They also showed significant desiccation tolerance, as indicated by the slower water loss in detached leaves. Our results indicate that the OsWRKY11 gene plays a role in heat and drought stress response and tolerance, and might be useful for improvement of stress tolerance.]]>
OsWRKY45 alleles play different roles in abscisic acid signalling and salt stress tolerance but similar roles in drought and cold tolerance in rice
DOI:10.1093/jxb/err144
URL
[本文引用: 1]
Although allelic diversity of genes has been shown to contribute to many phenotypic variations associated with different physiological processes in plants, information on allelic diversity of abiotic stress-responsive genes is limited. Here it is shown that the alleles OsWRKY45-1 and OsWRKY45-2 play different roles in abscisic acid (ABA) signalling and salt stress adaptation in rice. The two alleles had different transcriptional responses to ABA and salt stresses. OsWRKY45-1-overexpressing lines showed reduced ABA sensitivity, whereas OsWRKY45-1-knockout lines showed increased ABA sensitivity. OsWRKY45-1 transgenic plants showed no obvious difference from negative controls in response to salt stress. In contrast, OsWRKY45-2-overexpressing lines showed increased ABA sensitivity and reduced salt stress tolerance, and OsWRKY45-2-suppressing lines showed reduced ABA sensitivity and increased salt stress tolerance. OsWRKY45-1 and OsWRKY45-2 transgenic plants showed differential expression of a set of ABA- and abiotic stress-responsive genes, but they showed similar responses to cold and drought stresses. These results suggest that OsWRKY45-1 negatively and OsWRKY45-2 positively regulates ABA signalling and, in addition, OsWRKY45-2 but not OsWRKY45-1 negatively regulates rice response to salt stress. The different roles of the two alleles in ABA signalling and salt stress may be due to their transcriptional mediation of different signalling pathways.
OsWRKY30 is activated by MAP kinases to confer drought tolerance in rice
DOI:10.1007/s11103-012-9941-y
URL
[本文引用: 1]
Both the WRKY transcription factor (TF) and MAP kinases have been shown to regulate gene expression in response to biotic and abiotic stresses in plants. Several reports have shown that WRKY TFs may function downstream of MAPK cascades. Here, we have shown that OsWRKY30 interacted with OsMPK3, OsMPK4, OsMPK7, OsMPK14, OsMPK20-4, and OsMPK20-5, and could be phosphorylated by OsMPK3, OsMPK7, and OsMPK14. Overexpression of OsWRKY30 in rice dramatically increased drought tolerance. Overexpression of OsWRKY30AA, in which all SP (serine residue followed by proline residue) sites were replaced by AP (A, alanine), resulted in no improvement in drought tolerance. In addition, the function of transcriptional activation of OsWRKY30 was impaired after SP was replaced by AP. These results proved that the phosphorylation of OsWRKY30 by MAPKs was crucial in order for OsWRKY30 to perform its biological function.
The rice transcription factor OsWRKY47 is a positive regulator of the response to water deficit stress
DOI:10.1007/s11103-015-0329-7 URL [本文引用: 1]
Rice WRKY11 plays a role in pathogen defense and drought tolerance
DOI:10.1186/s12284-018-0199-0
URL
PMID:29330772
[本文引用: 1]
BACKGROUND: Plants are frequently subjected to abiotic and biotic stresses, and WRKY proteins play a pivotal role in the response to such stress. OsWRKY11 is induced by pathogens, drought, and heat, suggesting a function in biotic and abiotic stress responses. RESULTS: This study identified OsWRKY11, a member of WRKY group IIc. It is a transcriptional activator that localized to the nucleus. Ectopic expression of OsWRKY11 resulted in enhanced resistance to a bacterial pathogen, Xanthomonas oryzae pv. oryzae; resistance was compromised in transgenic lines under-expressing OsWRKY11. Ectopic expression of OsWRKY11 resulted in constitutive expression of defense-associated genes, whereas knock-down (kd) of OsWRKY11 reduced expression of defense-associated genes during pathogen attack, suggesting that OsWRKY11 activates defense responses. OsWRKY11 bound directly to the promoter of CHITINASE 2, a gene associated with defense, and activated its transcription. In addition, ectopic expression of OsWRKY11 enhanced tolerance to drought stress and induced constitutive expression of drought-responsive genes. Induction of drought-responsive genes was compromised in OsWRKY11-kd plants. OsWRKY11 also bound directly to the promoter of a drought-responsive gene, RAB21, activating its transcription. In addition, OsWRKY11 protein levels were controlled by the ubiquitin-proteasome system. CONCLUSION: OsWRKY11 integrates plant responses to pathogens and abiotic stresses by positively modulating the expression of biotic and abiotic stress-related genes.
Overexpression of a WRKY transcription factor Tawrky2 enhances drought stress tolerance in transgenic wheat
DOI:10.3389/fpls.2018.00997
URL
PMID:30131813
[本文引用: 1]
Drought is a major environmental stress that severely restricts plant growth and crop productivity. A previous study showed that TaWRKY2 from wheat (Triticum aestivum) plays an important role in drought stress tolerance. In the present study, we isolated the promoter of TaWRKY2 and identified multiple regulatory cis-elements in the promoter region. The activity of the TaWRKY2 promoter was induced by drought, salt, heat, and abscisic acid (ABA). We also generated TaWRKY2-overexpressing transgenic wheat, and found that the transgenic seedlings exhibited significantly enhanced tolerance to drought stress, as evidenced by a higher survival rate and lower water loss rate of detached leaves compared with wild type (WT) plants. In addition, the transgenic lines had higher contents of free proline, soluble sugar, and chlorophyll. During a prolonged period of drought stress before the heading stage, the growth of WT plants was inhibited, whereas the TaWRKY2-overexpressing lines progressed to the heading stage. The increased grain yield of the transgenic wheat lines reflected the cumulative effects of longer panicle length, more kernels per spike, and greater aboveground biomass. Our findings show that TaWRKY2 can enhance drought tolerance and increase grain yield in wheat, thus providing a promising candidate target for improving the drought tolerance of wheat cultivars through genetic engineering.
Maize WRKY transcription factor Zmwrky106 confers drought and heat tolerance in transgenic plants
DOI:10.3390/ijms19103046 URL [本文引用: 1]
Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans
DOI:10.1038/35888
URL
PMID:9486653
[本文引用: 1]
Experimental introduction of RNA into cells can be used in certain biological systems to interfere with the function of an endogenous gene. Such effects have been proposed to result from a simple antisense mechanism that depends on hybridization between the injected RNA and endogenous messenger RNA transcripts. RNA interference has been used in the nematode Caenorhabditis elegans to manipulate gene expression. Here we investigate the requirements for structure and delivery of the interfering RNA. To our surprise, we found that double-stranded RNA was substantially more effective at producing interference than was either strand individually. After injection into adult animals, purified single strands had at most a modest effect, whereas double-stranded mixtures caused potent and specific interference. The effects of this interference were evident in both the injected animals and their progeny. Only a few molecules of injected double-stranded RNA were required per affected cell, arguing against stochiometric interference with endogenous mRNA and suggesting that there could be a catalytic or amplification component in the interference process.
ThNAC13,a NAC transcription factor from Tamarix hispida,confers salt and osmotic stress tolerance to transgenic Tamarix and Arabidopsis
DOI:10.3389/fpls.2017.00635
URL
PMID:28491072
[本文引用: 1]
NAC (NAM, ATAF1/2, and CUC2) proteins play critical roles in many plant biological processes and environmental stress. However, NAC proteins from Tamarix hispida have not been functionally characterized. Here, we studied a NAC gene from T. hispida, ThNAC13, in response to salt and osmotic stresses. ThNAC13 is a nuclear protein with a C-terminal transactivation domain. ThNAC13 can bind to NAC recognized sites and calmodulin-binding NAC (CBNAC) binding element. Overexpression of ThNAC13 in Arabidopsis improved seed germination rate and increased root growth and fresh weight gain under salt or osmotic stress. Transgenic T. hispida plants transiently overexpressing ThNAC13 and with RNAi-silenced ThNAC13 were generated for gain- and loss-of-function experiments. Following exposure to salt or osmotic stress, overexpression of ThNAC13 induced superoxide dismutase (SOD) and peroxidase (POD) activities, chlorophyll and proline contents; decreased the reactive oxygen species (ROS) and malondialdehyde levels; and reduced electrolyte leakage rates in both transgenic Tamarix and Arabidopsis plants. In contrast, RNAi-silenced ThNAC13 showed the opposite results in transgenic Tamarix. Furthermore, ThNAC13 induced the expression of SODs and PODs in transgenic Arabidopsis. These results suggest that ThNAC13 improves salt and osmotic tolerance by enhancing the ROS-scavenging capability and adjusting osmotic potential.
Two insulin receptors determine alternative wing morphs in planthoppers
DOI:10.1038/nature14286
URL
PMID:25799997
[本文引用: 1]
Wing polyphenism is an evolutionarily successful feature found in a wide range of insects. Long-winged morphs can fly, which allows them to escape adverse habitats and track changing resources, whereas short-winged morphs are flightless, but usually possess higher fecundity than the winged morphs. Studies on aphids, crickets and planthoppers have revealed that alternative wing morphs develop in response to various environmental cues, and that the response to these cues may be mediated by developmental hormones, although research in this area has yielded equivocal and conflicting results about exactly which hormones are involved. As it stands, the molecular mechanism underlying wing morph determination in insects has remained elusive. Here we show that two insulin receptors in the migratory brown planthopper Nilaparvata lugens, InR1 and InR2, have opposing roles in controlling long wing versus short wing development by regulating the activity of the forkhead transcription factor Foxo. InR1, acting via the phosphatidylinositol-3-OH kinase (PI(3)K)-protein kinase B (Akt) signalling cascade, leads to the long-winged morph if active and the short-winged morph if inactive. InR2, by contrast, functions as a negative regulator of the InR1-PI(3)K-Akt pathway: suppression of InR2 results in development of the long-winged morph. The brain-secreted ligand Ilp3 triggers development of long-winged morphs. Our findings provide the first evidence of a molecular basis for the regulation of wing polyphenism in insects, and they are also the first demonstration--to our knowledge--of binary control over alternative developmental outcomes, and thus deepen our understanding of the development and evolution of phenotypic plasticity.
Knockdown of five trehalase genes using RNA interference regulates the gene expression of the chitin biosynthesis pathway in Tribolium castaneum
DOI:10.1186/s12896-016-0297-2
URL
PMID:27596613
[本文引用: 1]
BACKGROUND: RNA interference is a very effective approach for studies on gene function and may be an efficient method for controlling pests. Trehalase is a key gene in the chitin biosynthesis pathway in insects. Five trehalase genes have been cloned in Tribolium castaneum, though it is not known whether the detailed functions of these trehalases can be targeted for pest control. RESULTS: The functions of all five trehalase genes were studied using RNAi, and the most important results showed that the expression of all 12 genes decreased significantly from 12 to 72 h compared with the control groups, except GP1 at 72 h, when the expression of the TcTre2 gene was suppressed. The results also revealed different abnormal phenotypes, and the observed mortality rates ranged from 17 to 42 %. The qRT-PCR results showed that the expression of TPS, GS, two GP, CHS1a and CHS1b genes decreased significantly, while that of the CHS2 gene decreased or increased after RNAi after the five trehalases were silenced at 48 h. In addition, TPS gene expression decreased from 12 to 72 h after dsTcTre injection. CONCLUSIONS: These results demonstrate that silencing of any individual trehalase gene, especially Tre1-4 and Tre2 gene can lead to moulting deformities and a high mortality rate through the regulation of gene expression in the chitin biosynthesis pathway and may be a potential approach for pest control in the future.
Identification of a novel cytochrome P450 gene,CYP321E1 from the diamondback moth,Plutella xylostella (L.) and RNA interference to evaluate its role in chlorantraniliprole resistance
DOI:10.1017/S0007485314000510
URL
[本文引用: 1]
Insect cytochrome P450 monooxygenases (P450s) play an important role in catalysis of many reactions leading to insecticides resistance. Our previous studies on transcriptome analysis of chlorantraniliprole-resistant development in the diamondback moth, Plutella xylostella revealed that up-regulation of cytochrome P450s are one of the main factors leading to the development of chlorantraniliprole resistance. Here, we report for the first time a novel cytochrome P450 gene CYP321E1, which belongs to the cytochrome P450 gene family CYP321. Real-time quantitative PCR (RT-qPCR) analyses indicated that CYP321E1 was expressed at all developmental stages of P. xylostella but was highest in the fourth-instar larvae; furthermore, the relatively high expression was observed in the midgut of the fourth-instar larvae, followed by fat bodies and epidermis. The expression of CYP321E1 in P. xylostella was differentially affected by three representative insecticides, including alphamethrin, abamectin and chlorantraniliprole. Among them, the exposure to chlorantraniliprole resulted in the largest transcript level of this cytochrome P450 gene. The findings suggested potential involvement of CYP321E1 in chlorantraniliprole resistance of P. xylostella. To assess the functional link of CYP321E1 to chlorantraniliprole resistance, RNA interference (RNAi)-mediated gene silencing by double stranded RNA (dsRNA) injecting was used. Results revealed that injection delivery of dsRNA can greatly reduce gene expression after 24 h. As a consequence of RNAi, a significant increment in mortality of larvae injected CYP321E1 dsRNA was observed after 24h of exposure to chlorantraniliprole. These results strongly support our notion that this novel cytochrome P450 gene plays an important role in chlorantraniliprole detoxification in the diamondback moth and is partly responsible for its resistance.
The novel ABC transporter ABCH1 isa potential target for RNAi-based insect pest control and resistance management
DOI:10.1038/srep13728
URL
PMID:26333918
[本文引用: 1]
Insect pests cause serious crop damage and develop high-level resistance to chemical insecticides and Bacillus thuringiensis (Bt) insecticidal Cry toxins. A new promising approach for controlling them and overcoming this resistance is RNA interference (RNAi). The RNAi-based insect control strategy depends on the selection of suitable target genes. In this study, we cloned and characterized a novel ABC transporter gene PxABCH1 in diamondback moth, Plutella xylostella (L.). Phylogenetic analysis showed that PxABCH1 is closely related to ABCA and ABCG subfamily members. Spatial-temporal expression detection revealed that PxABCH1 was expressed in all tissues and developmental stages, and highest expressed in head and male adult. Midgut sequence variation and expression analyses of PxABCH1 in all the susceptible and Bt-resistant P. xylostella strains and the functional analysis by sublethal RNAi demonstrated that Cry1Ac resistance was independent of this gene. Silencing of PxABCH1 by a relatively high dose of dsRNA dramatically reduced its expression and resulted in larval and pupal lethal phenotypes in both susceptible and Cry1Ac-resistant P. xylostella strains. To our knowledge, this study provides the first insight into ABCH1 in lepidopterans and reveals it as an excellent target for RNAi-based insect pest control and resistance management.
RNAi-based inhibition of porcine reproductive and respiratorysyndrome virus replication intransgenic pigs
DOI:10.1016/j.jbiotec.2013.11.022
URL
[本文引用: 1]
Porcine reproductive and respiratory syndrome (PRRS) is an economically devastating viral disease causing heavy losses to the swine industry worldwide. Many studies have shown that transient delivery of small interfering RNA ( siRNA) or adenovirus-mediated RNA interfere (RNAi) could potentially inhibit porcine reproductive and respiratory syndrome virus (PRRSV) replication in vivo and in vitro. Here, we applied RNAi to produce transgenic (TG) pigs that constitutively expressed PRRSV-specific siRNA derived from small hairpin RNA ( shRNA). First, we evaluated siRNA expression in the founding and Fl generation pigs and confirmed stable transmission. Then, we detected the expression of IFN-p and protein kinase R (PKR) and found no difference among TG, non-transgenic (NTG), and wild-type pigs. Lastly, the Fl generation pigs, including TG and NTG piglets, were challenged with 3 x 104.5 TCID50 of JXA1, a highly pathogenic porcine reproductive and respiratory syndrome virus (HP-PRRSV). Our results showed that the in vivo siRNA expression substantially reduced the serum HP-PRRSV titers and increased survival time by 3 days when TG pigs were compared with the NTG controls. These data suggested that RNAi-based genetic modification might be used to breed viral-resistant livestock with stable siRNA expression with no complications of siRNA toxicity. (C) 2013 Published by Elsevier B.V.
Inhibiting avian influenza virus shedding using a novel RNAi antiviral vector technology:proof of concept inan avian cell model
DOI:10.1186/s13568-016-0187-y
URL
PMID:26910902
[本文引用: 1]
Influenza A viruses pose significant health and economic threats to humans and animals. Outbreaks of avian influenza virus (AIV) are a liability to the poultry industry and increase the risk for transmission to humans. There are limitations to using the AIV vaccine in poultry, creating barriers to controlling outbreaks and a need for alternative effective control measures. Application of RNA interference (RNAi) techniques hold potential; however, the delivery of RNAi-mediating agents is a well-known obstacle to harnessing its clinical application. We introduce a novel antiviral approach using bacterial vectors that target avian mucosal epithelial cells and deliver (small interfering RNA) siRNAs against two AIV genes, nucleoprotein (NP) and polymerase acidic protein (PA). Using a red fluorescent reporter, we first demonstrated vector delivery and intracellular expression in avian epithelial cells. Subsequently, we demonstrated significant reductions in AIV shedding when applying these anti-AIV vectors prophylactically. These antiviral vectors provided up to a 10,000-fold reduction in viral titers shed, demonstrating in vitro proof-of-concept for using these novel anti-AIV vectors to inhibit AIV shedding. Our results indicate this siRNA vector technology could represent a scalable and clinically applicable antiviral technology for avian and human influenza and a prototype for RNAi-based vectors against other viruses.
PIK3CA and PIK3CB silencing by RNAi reverse MDR and inhibit tumorigenic properties in human colorectal carcinoma
DOI:10.1007/s13277-015-4691-5
URL
PMID:26747178
[本文引用: 1]
Colorectal carcinoma (CRC) is the second most common and frequent cause of cancer-related deaths for men and women in the world. PIK3CA and PIK3CB that reverse multidrug resistance (MDR) can serve as predictive and prognostic markers as well as therapeutic targets for CRC treatment. In the present study, we showed that PIK3CA and PIK3CB are upregulated in CRCs and positively correlated with MDR-1, LRP, and GST-pi. Long-term monitoring of 316 CRC patients showed that PIK3CA and PIK3CB were associated with poor survival time as shown by Kaplan-Meier analysis. Furthermore, we found that the downregulation of PIK3CA and PIK3CB reversed MDR; inhibited the capability of proliferation, migration, and invasion of CRC cells; and slowed down the CRC tumor growth in nude mice. Consistent with clinical observations, PIK3CA and PIK3CB significantly increase multidrug resistance of CRC cells in vivo. Together, these results suggest that PIK3CA and PIK3CB may be used as potential therapeutic drug targets for colorectal cancer.
Lentivirus-mediated RNAi knockdown of LMP2A inhibits the growth of the Epstein-Barrassociated gastric carcinoma cell line GT38 in vitro
DOI:10.3892/etm.2016.3954
URL
PMID:28123488
[本文引用: 1]
In this study, lentivirus-mediated RNA interference (RNAi) was applied to inhibit latent membrane protein 2A (LMP2A) gene expression, in order to explore the effects of LMP2A silencing on the growth of an Epstein-Barr virus-associated gastric carcinoma (EBVaGC) cell line in vitro. Lentivirus-mediated RNAi technology was employed to specifically knock down the LMP2A gene in the EBV-positive gastric carcinoma cell line GT38. After infection, reverse transcription-quantitative polymerase chain reaction, western blotting, flow cytometry and colony formation assays were conducted to evaluate the expression of LMP2A and the biological behavior of the GT38 cell line in vitro. The results showed that the expression of the LMP2A gene was clearly downregulated in the infected cells, which indicated that a highly efficient and stable lentivirus vector was successfully constructed. In the GT38 cells in which the expression of LMP2A was downregulated, the proliferation and colony formation of the cells was significantly inhibited. In addition, it was found that the cell cycle of the GT38 cells was arrested in the G0/G1 phase and the apoptosis rate was increased. These results indicate that lentivirus-mediated RNAi knockdown of LMP2A inhibits the growth of the EBVaGC cell line GT38 in vitro, and suggests that LMP2A is a potential target for gene therapy in the treatment of EBVaGC.
Harnessing RNAi-based nanomedicines for therapeutic gene silencing in B-cell malignancies
玉米淀粉分支酶基因反义表达载体的构建和功能分析
[1,2]。目前,对淀粉分支酶及其基因已从分子水平和生物化学方面进行了一些研究,淀粉分支酶基因的生理功能基本清楚[3~5]。由于反义RNA(antiscnse RNA, as RNA)技术提供了直接有效地人为控制基因表达的方法,现已成为基因工程研究中的一项重要技术[6]。本项研究利用基因工程技术,克隆玉米淀汾分支酶基因,构建反义表达载体。通过花粉管通道法将其导入玉米自交系,旨在抑制淀粉分支酶基因的表达,提高直链淀粉的含量。]]>
A versatile zero background T-vector system for gene cloning and functional genomics1 [C][W][OA]
Highly specific gene silencing by artificial microRNAs in Arabidopsis
DOI:10.1105/tpc.105.039834
URL
PMID:16531494
[本文引用: 1]
Plant microRNAs (miRNAs) affect only a small number of targets with high sequence complementarity, while animal miRNAs usually have hundreds of targets with limited complementarity. We used artificial miRNAs (amiRNAs) to determine whether the narrow action spectrum of natural plant miRNAs reflects only intrinsic properties of the plant miRNA machinery or whether it is also due to past selection against natural miRNAs with broader specificity. amiRNAs were designed to target individual genes or groups of endogenous genes. Like natural miRNAs, they had varying numbers of target mismatches. Previously determined parameters of target selection for natural miRNAs could accurately predict direct targets of amiRNAs. The specificity of amiRNAs, as deduced from genome-wide expression profiling, was as high as that of natural plant miRNAs, supporting the notion that extensive base pairing with targets is required for plant miRNA function. amiRNAs make an effective tool for specific gene silencing in plants, especially when several related, but not identical, target genes need to be downregulated. We demonstrate that amiRNAs are also active when expressed under tissue-specific or inducible promoters, with limited nonautonomous effects. The design principles for amiRNAs have been generalized and integrated into a Web-based tool (http://wmd.weigelworld.org).
转Bt基因玉米幼苗残体中CrylAb杀虫蛋白田间降解动态
【目的】研究间苗后留在田间地表的转Bt基因玉米幼苗残体中Cry1Ab杀虫蛋白的降解规律,比较两种Bt玉米幼苗残体中Cry1Ab杀虫蛋白的降解速度。【方法】以两种表达Cry1Ab杀虫蛋白的转Bt基因抗虫玉米MON810和Bt11为材料,采用ELISA方法测定各取样时期中幼苗残体中Cry1Ab杀虫蛋白残留量。【结果】转Bt基因玉米幼苗残体中杀虫蛋白降解是逐渐的,且降解速度较快,到50 d时幼苗残体已经完全腐烂,Bt11幼苗残体中的杀虫蛋白已经完全降解,在MON810中还能检测到微量的杀虫蛋白。两种转基因玉米幼苗残体中的Bt杀虫蛋白的初始含量差异不显著,但在同一时间段的Bt杀虫蛋白降解速度存在差异均显著,在30 d前MON810幼苗残体中Bt杀虫蛋白降解速度比Bt11降解的快,30d后,则降解趋势相反,到50 d取样结束时MON810和Bt11分别降解了初始含量的99.81%和100%。【结论】两种转Bt基因玉米间苗后留在田间的幼苗残体中的Cry1Ab杀虫蛋白降解速度不同,在50 d完全腐烂时,其中的杀虫蛋白完全降解或仅有微量残留。
大豆胰蛋白酶抑制剂KSTI3基因的克隆及其植物表达载体的构建
KSTI3的全长DNA片段,并将其构建到pMD18-T vector上。核苷酸序列测定结果表明:该基因片段全长654 bp,与已发表的KSTI3基因序列同源性达99%。将反义+正义基因片段插入到pBI121 35S启动子下,构建重组质粒pBIKSTI3。通过冻融法将该重组质粒转入根癌农杆菌EHA105中,获得了植物表达载体。]]>
One-step DNA fragment assembly for expressing intron-containing hairpin RNA in plants for gene silencing
DOI:10.1016/j.ab.2012.09.034
URL
[本文引用: 1]
Double-stranded RNA-mediated RNA interference in plants involves generating a construct expressing intron-containing hairpin RNA (ihpRNA), which usually is a cumbersome, multistep process. Here, we describe a simplified method involving single steps of PCR, restriction, ligation, and transformation for assembling an ihpRNA construct for plant transformation. Our method has several advantages over the currently available ones, viz., wider choice of restriction sites and facility for rapid screening of positive clones, among others. We demonstrate the utility of this approach in assembling the tomato phytoene desaturase gene. This simplified DNA fragment assembly strategy for ihpRNA construction facilitates high-throughput gene silencing in plants. (C) 2012 Elsevier Inc.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}