


开放科学(资源服务)标识码(OSID): 
目前,国内外利用不同遗传背景的群体,采用连锁分析的方法,在1~10染色体上均发现了控制玉米秸秆IVDMD的QTL[5,6,7,8]。全基因组关联分析(genome-wide association study,GWAS)具有高分辨率且省时等特点,是检测复杂性状变异强大的遗传方法,特别是玉米作为异交作物,重组较多,导致较短的连锁不平衡(linkage disequilibrium,LD),且表型和遗传多样性丰富,分辨率较高[9,10,11],但不同群体LD存在差异,如农家种1kb、广泛变异群体1.5kb、优良自交系100kb,都利于关联定位的精度[10]。目前关于青贮玉米秸秆消化率性状的全基因组关联分析的研究极少。Wang等[9]以559 285个高质量SNPs对368份玉米自交系采用GWAS方法鉴定了82个秸秆IVDMD显著相关的位点,并发现了直接参与细胞壁生物合成的基因ZmC3H2。但Wang等[9]利用关联分析对秸秆IVDMD进行研究时,群体中没有青贮玉米品种的亲本自交系,从而不利于挖掘更多且直接的优良的青贮玉米消化率性状遗传位点。
本研究以341份玉米自交系为材料,构建关联作图群体,基于全基因组测序的高质量SNPs进行秸秆48h IVDMD的全基因组关联分析,挖掘优良等位变异,筛选候选基因,为优质青贮玉米选育提供参考。
1 材料与方法
1.1 供试材料及田间设计
供试材料为194份美国玉米自交系和147份国内玉米自交系(包括14份国内常用玉米自交系和133份课题组自选玉米自交系)构成的341份关联群体,其中6份为青贮玉米品种的亲本自交系。
2018年5月初将供试材料分别种植于辽宁省沈阳市东亚国际种业公司和内蒙古自治区通辽市农业科学研究院科学研究基地。行长5.0m,行距0.6m,株距0.25m,双行区,自然散粉,田间管理同一般玉米自交系试验田。
1.2 品质测定方法
调查供试材料生育时期(主要包括抽雄期和吐丝期)。在玉米籽粒乳线位置1/2时,选取长势一致的10株玉米,从地上部20cm处刈割,去掉果穗,收获秸秆。用风送式动力铡草机粉碎后混合均匀,平行取两份1 000g装入布袋,立刻放入105℃烘箱中杀青2h,散热散水气后调至65℃烘干至恒重,用锤片式粉碎机磨碎,过40目筛,装入牛皮纸袋,于65℃烘箱烘至恒重,取出样品冷却至室温,待用。
试验采用FOSS近红外光谱仪(NIRS DS2500)[12]对供试样品48h IVDMD(%)进行测定,该仪器采用前分光单色仪技术,探测器是硅(400~1 100nm)和硫化铅(1 100~2 500nm),光谱分辨率是0.5nm-1。将每份样品混匀,取适量于直径为50mm的石英柱形样品杯中,波长为400~2 500nm近红外光以漫反射形式从样杯底部扫描,每份样品扫描2次,每次重复重新取样品,结果取均值(所测定含量是干基)。
1.3 表型统计方法
采用SPSS 20.0进行描述性统计分析和相关性分析,利用R 3.5.1软件对表型频率分布作图,利用Microsoft Excel 2010进行双因素方差分析估算广义遗传力(HB2),HB2=VG/VP×100%=VG/(VG+VE)×100%,VP、VG和VE分别是表型变异方差、遗传变异方差和环境变异方差。
1.4 基因型测定及分析
1.5 群体结构和亲缘关系
采用fastStructure 1.0软件[15],以6 276 612个MAF>0.05且均匀分布染色体的SNPs,对341份供试材料进行贝叶斯聚类,计算不同层次的亚群数(K=2~10),再根据最大似然数LnP(D),确定本群体理论上由5大亚群组成。
1.6 秸秆48h IVDMD全基因组关联分析
1.7 秸秆48h IVDMD定位结果比较分析
借助前人用不同群体构建遗传连锁图谱进行的QTL定位结果,若本试验关联定位结果落入前人QTL定位区间内或相差200kb以内说明本试验关联定位效果良好。
1.8 秸秆48h IVDMD候选基因预测
1.9 群体变异位点分析
2 结果与分析
2.1 秸秆48h IVDMD表型分析
图1
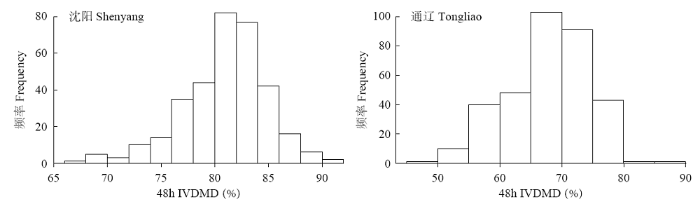
表1 两个环境秸秆48h IVDMD统计分析
Table 1
| 地点 Location | 均值 Mean (%) | 变异范围 Range (%) | 变异系数 Coefficient of variation (%) | 偏度 Skewness (%) | 峰度 Kurtosis (%) | 相关系数 Correlation coefficient | 广义遗传力 Broad-sense heritability (%) |
|---|---|---|---|---|---|---|---|
| 沈阳Shenyang | |||||||
| 沈阳Shenyang | 80.96 | 67.24~91.41 | 4.85 | -0.64 | 1.04 | ||
| 通辽Tongliao | 67.94 | 49.75~85.94 | 9.50 | -0.40 | -0.24 | 0.63** | 71.98 |
注:**在0.01水平上显著相关
Note: ** Significant correlation at 0.01 level
2.2 秸秆48h IVDMD全基因组关联分析
利用FarmCPU模型对341份玉米自交系的48h IVDMD进行全基因组关联分析。绘制曼哈顿图(Manhattan)(图2),超过水平线(两条水平线代表全基因组显著水平阈值6.0和8.0)的SNPs即为与本试验目标性状显著相关的位点。两个环境中FarmCPU模型的QQ图表示群体结构较为良好地被控制(图3)。在沈阳和通辽的两个环境中共检测到153个显著SNPs位点(P<1.0×10-6),分布在玉米第1~10染色体,其中,沈阳环境中,在显著水平P<1.0×10-8上检测到4个显著SNPs位点,分布于第2、3、6、8染色体上(图4)。在第1~3、6~8、10染色体上,沈阳和通辽两个环境中都检测出了显著SNPs位点(P<1.0×10-6),但未发现相同的显著SNPs位点,位于10号染色体的chr10_131199866标记(沈阳)和chr10_130000984、chr10_130001456、chr10_130001612、chr10_130001742、chr10_130001760 5个连锁标记(通辽)最近,距离是1.20Mb。
图2
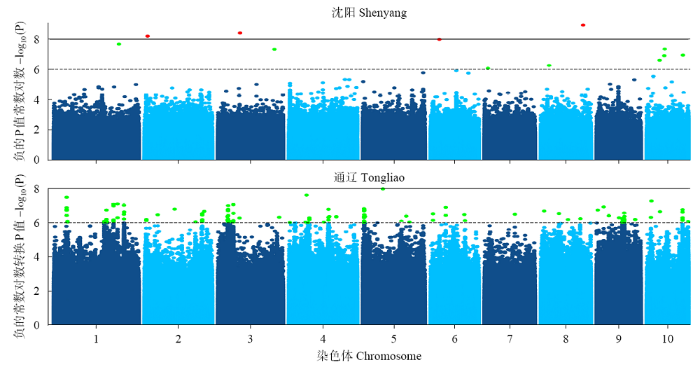
图2
不同环境(沈阳和通辽)秸秆48h IVDMD全基因组关联曼哈顿图
实线和虚线分别代表显著阈值8.0和6.0
Fig.2
Manhattan plot of genome-wide association under the environments of Shenyang and Tongliao
The solid and dashed lines represent significance thresholds 8.0 and 6.0, respectively
图3
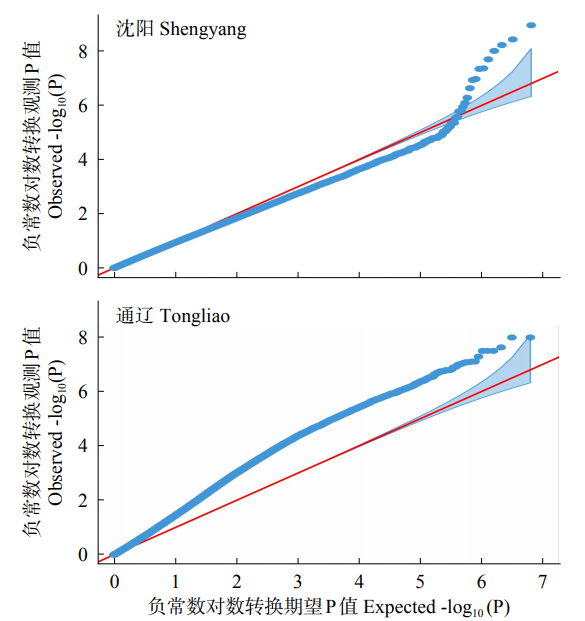
图3
不同环境秸秆48h IVDMD全基因组关联QQ图
红线表示零假设预期分布,蓝线表示观察到的和通辽秸秆48h IVDMD百分比的关联分布
Fig.3
Quantile-quantile plot of genome-wide association under the environments of Shenyang and Tongliao
The red line represents the expected distribution of null hypothesis, and the blue line represents the observed correlation distribution with the percentage of Tongliao straw at 48h IVDMD
图4
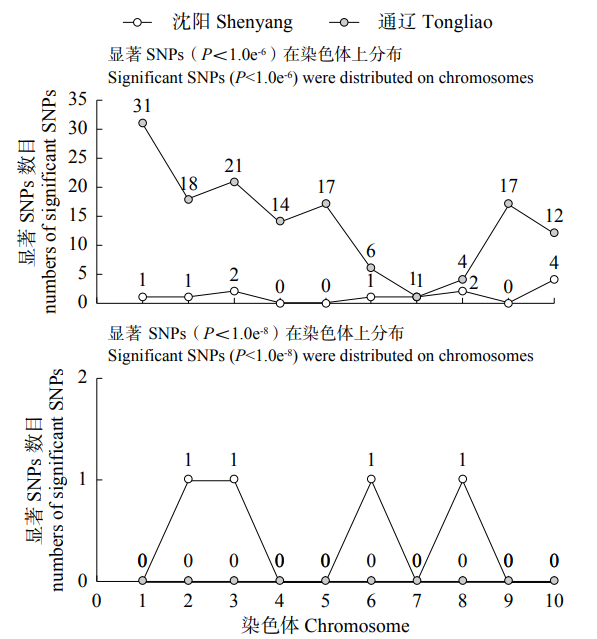
采用混合线性模型(MLM)对341份玉米自交系的48h IVDMD进行全基因组关联,共发现分布在第2、4~9染色体的10个SNPs显著位点(P<1.0×10-6),结果仍没有发现两个环境相同的SNPs显著位点(P<1.0×10-6)(图5)。利用MLM不仅没有关联到两个环境中和两个性状显著相关的相同的位点,而且两个环境中在共同染色体上也没有检测到显著位点。从两个模型的曼哈顿图和QQ图可以看出,FarmCPU模型关联的显著位点优于MLM的,说明FarmCPU模型关联效率要高于MLM,候选基因易从FarmCPU模型关联的显著位点中预测。
图5
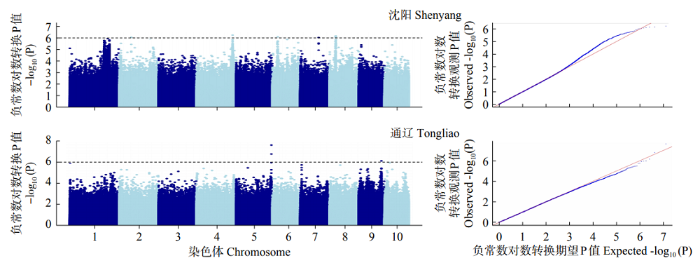
图5
两个环境秸秆48h IVDMD全基因组关联曼哈顿图及其QQ图(MLM)
Fig.5
Manhattan plots and its QQ plots of genome-wide association in two environments (MLM)
2.3 候选基因分析
图6
3 讨论
3.1 GWAS定位结果分析
Li等[5]以郑58和HD568组成的220份RIL群体为材料,用6个消化性状定位了47个QTLs,其中位于2、7~10号染色体的9个QTLs控制IVDMD,解释单个QTL 5.3%的表型变异,其中定位在9.03染色体上QTL和Chr9_25266410标记最小仅距88kb。Wang等[7]以CE03005高油玉米自交系和B73组成的211份材料对玉米消化性状进行QTL研究,其中,在4~6和10号染色体上发现有6个位点与IVDMD显著相关,解释单个位点10.6%的表型变异。本试验和48h IVDMD显著相关的标记chr4_59965077、chr4_63943611、chr4_69161939、chr4_74067130、chr4_74067534和chr4_74243482均定位在了bin4.05染色体区段内,本试验和48h IVDMD显著相关的标记chr6_31376191、chr6_8473014、chr6_8473017、chr6_54601996和chr6_54602105均定位在了bin6.01染色体区段内,本试验和48h IVDMD显著相关的标记chr10_46816004、ch10_63652102、chr10_65363792、chr10_15312554、chr10_17307934和chr10_48100033均定位在了bin10.03染色体区段内,与本研究结果基本一致。王琪[6]用B73和By804作亲本构建RIL群体,在3号染色体鉴定了消化率的遗传位点,本试验在这个区域也发现了相关QTL。
本试验并非所有显著SNP位点都在已知的QTL定位区间,可能原因在于:QTL作图受亲本遗传背景或检测微效QTL功效低的原因;双亲群体中的功能位点可能在关联分析群体中是稀有等位变异,因而导致在关联分析群体中不能检测;关联分析群体存在双亲群体中可能不存在的遗传变异;关于IVDMD的QTL研究非常少见。
3.2 有潜力的候选基因分析
鉴定玉米秸秆消化率的相关基因能更好地理解细胞壁合成的分子遗传机制。在本试验38个候选基因中,基因Zm00001d009009编码产物类糖基转移酶KOBITO1是高度保守的、具有植物特异性的新型质膜结合蛋白,该蛋白是拟南芥细胞扩张过程中合成纤维素所必需的蛋白。在所有光和黑暗环境中,KOB1基因突变导致拟南芥下胚轴变短,与野生型植株相比,该突变体中纤维素的相对含量下降了33%,证明该蛋白通过调节纤维素合成,促进植株下胚轴生长[26,27]。基因Zm00001d028874编码产物是木葡聚糖6-木糖基转移酶2,木葡聚糖是禾本科植物初生细胞壁半纤维素合成的主要组分,木糖基转移酶2(XXT2)将核苷酸二磷酸糖中的木糖残基转移到木聚糖主链中的葡聚糖O-6位置,研究表明,XXT2突变使木聚糖含量下降近30%,另外,木葡聚糖6-木糖基转移酶突变导致水稻根毛发育异常;有研究表明,编码木葡聚糖6-木糖基转移酶基因OsXXT1在维持水稻细胞壁结构和抗拉强度方面很重要[28,29,30]。
秸秆细胞壁相关性状不仅能调节玉米生长,而且在作物育种上,与抗逆性表现出很强的相关性[31],可以增强对病虫的免疫机能,提高对不利环境的适应能力。在本试验38个候选基因中,基因Zm00001d031449含有1个非同义SNP,将333位和678位的异亮氨酸突变成甲硫氨酸,编码叶绿体脂氧合酶Ⅱ。虫害的番茄叶片中TomLoxD基因上调,编码1个叶绿体LOX,该LOX可能是十八烷类防御信号通路的组成部分,该通路在一系列酶的作用下产生的茉莉酸与其受体结合,活化相应基因,产生能削弱或阻断害虫消化道内蛋白酶的蛋白酶抑制素,使害虫厌食或消化不良而死[32,33]。Zm00001d044201含有1个非同义SNP,将221位的丙氨酸突变成丝氨酸,编码ATP依赖Zn2+蛋白酶FTSH11,仅位于叶绿体和线粒体,研究表明,在高于30℃的环境中,拟南芥FtSH11突变体的生长发育受到抑制,而野生型植株不受影响,故该基因产物可能和植物耐热相关[34]。基因Zm00001d031449和Zm00001d044201可能因自然选择而发生非同义突变,但编码产物对植物生长和适应性存在正调节作用。基因Zm00001d043581编码产物是一类类枯草杆菌蛋白酶,与病原体识别及互作,同时被病原体诱导,启动细胞程序性死亡和超敏反应,对植物产生免疫反应[35]。综上,这些抗性基因的发现说明秸秆细胞壁是保护植株免受生物和非生物伤害的天然屏障。
综上,玉米秸秆细胞壁是玉米健壮生长的必需结构,也是草食牲畜主要的纤维和能量来源,理解秸秆细胞壁合成机制和玉米生长适应性机制对找到有关消化率性状的基因有帮助。本文初探了青贮玉米秸秆48h IVDMD的遗传机制,为分子改良纤维含量和消化率提供了数据支撑,也为优质青贮玉米选育提供了理论参考。
4 结论
利用6 276 612个高质量SNPs(MAF>0.05)对341份玉米自交系秸秆48h IVDMD进行全基因组关联分析,两个环境中采用FarmCPU模型在所有染色体上检测到153个SNPs位点(P<1.0×10-6)和秸秆48h IVDMD显著相关,共挖掘38个候选基因,主要涉及细胞生长发育、防御反应、信号转导和细胞代谢等相关生物学功能。Zm00001d009009基因和Zm00001d028874基因可能分别与玉米秸秆纤维合成和初生壁合成有关。
参考文献
Genetic variation and breeding strategies for improved cell wall digestibility in annual forage crops. A review
Genetic and quantitative trait locus analysis of cell wall components and forage digestibility in the Zheng58 × HD568 maize RIL population at anthesis stage
DOI:10.3389/fpls.2017.01472
URL
PMID:28883827
[本文引用: 2]

10% of the phenotypic variation in the RIL population, whereas 70.2% (33/47) explained
Genetic analysis and QTL mapping of stalk digestibility and kernel composition in a high-oil maize mutant (Zea mays L.)
Genetic analysis and QTL mapping of cell wall digestibility and lignification in silage maize
Genome-wide association analysis of forage quality in maize mature stalk
Natural variations and genome-wide association studies in crop plants. annual. review
可见/近红外光谱分析秸秆-煤混燃物的秸秆含量
快速检测秸秆-煤混燃物对生物质混燃发电中补贴政策的制定具有重要意义。该研究采用可见/近红外光谱法定性判别秸秆、煤和秸秆-煤混燃物,定量分析秸秆-煤混燃物中秸秆含量。收集并制备秸秆样品80个(粒径小于80 mm)、煤样品9个(粒径小于10 mm),制备秸秆质量分数为70%~99%的秸秆-煤混燃物样品120个(混燃物1)、秸秆分数含量为1%~30%的秸秆-煤混燃物样品120个(混燃物2)。使用FOSS NIRS DS 2500型光谱仪获取样品光谱。分别使用偏最小二乘判别法(PLS-DA)建立定性分析模型,使用改进的偏最小二乘法(MPLS)建立定量分析模型。结果显示,在秸秆和混燃物1之间进行判别,使用1100~2 500 nm谱区,正确判别率为90.00%;在煤和混燃物2之间进行判别,使用400~2 500 nm谱区,正确判别率为71.88%;定量分析混燃物1和混燃物2中秸秆含量,相对分析误差分别为2.32(400~2 500 nm谱区)和1.48(400~1 100 nm谱区)。研究结果表明,1 100~2 500 nm谱区较适合秸秆和混燃物1之间的判别,该谱区同样适合定量分析混燃物1中秸秆含量。400~1 100 nm谱区较适合煤和混燃物2之间的判别,该谱区同样适合定量分析混燃物2中秸秆含量。可见/近红外光谱结合化学计量学是快速定性和定量分析大粒度秸秆-煤混燃物的可行方法。
玉米基因组DNA提取及浓度测定方法评价
以非转基因玉米种子为材料,比较了常用的3种植物基因组DNA提取试剂盒及改良的CTAB法,通过琼脂糖凝胶电泳、紫外分光光度及实时荧光PCR扩增检测,对提取得到的基因组DNA的纯度、得率及4种提取方法的重复性、提取时间进行分析;比较紫外分光光度法、Qubit荧光法、Pico Green荧光分光光度法,以实时荧光定量PCR检测结果为参照,对3种DNA浓度测定方法的准确性进行分析。结果显示,磁珠法(Promega)最适合应用于快速、简便、高效检测中的植物基因组DNA提取,能有效获得纯度高、完整性好的基因组DNA,并且磁珠法提取效率高,重复性好,提取时间短;在基因组DNA浓度测定中,紫外分光光度法、Qubit荧光法、Pico Green荧光分光光度法的相对误差分别为99.8%、49.8%和28.9%,表明Pico Green荧光分光光度法测定DNA浓度的准确度最高。
Fast and accurate short read alignment with Burrows-Wheeler transform
DOI:10.1093/bioinformatics/btp324
URL
PMID:19451168
[本文引用: 1]
MOTIVATION: The enormous amount of short reads generated by the new DNA sequencing technologies call for the development of fast and accurate read alignment programs. A first generation of hash table-based methods has been developed, including MAQ, which is accurate, feature rich and fast enough to align short reads from a single individual. However, MAQ does not support gapped alignment for single-end reads, which makes it unsuitable for alignment of longer reads where indels may occur frequently. The speed of MAQ is also a concern when the alignment is scaled up to the resequencing of hundreds of individuals. RESULTS: We implemented Burrows-Wheeler Alignment tool (BWA), a new read alignment package that is based on backward search with Burrows-Wheeler Transform (BWT), to efficiently align short sequencing reads against a large reference sequence such as the human genome, allowing mismatches and gaps. BWA supports both base space reads, e.g. from Illumina sequencing machines, and color space reads from AB SOLiD machines. Evaluations on both simulated and real data suggest that BWA is approximately 10-20x faster than MAQ, while achieving similar accuracy. In addition, BWA outputs alignment in the new standard SAM (Sequence Alignment/Map) format. Variant calling and other downstream analyses after the alignment can be achieved with the open source SAMtools software package. AVAILABILITY: http://maq.sourceforge.net.
FastSTRUCTURE:variational inference of population structure in large SNP data sets
DOI:10.1534/genetics.114.164350
URL
[本文引用: 1]
Tools for estimating population structure from genetic data are now used in a wide variety of applications in population genetics. However, inferring population structure in large modern data sets imposes severe computational challenges. Here, we develop efficient algorithms for approximate inference of the model underlying the STRUCTURE program using a variational Bayesian framework. Variational methods pose the problem of computing relevant posterior distributions as an optimization problem, allowing us to build on recent advances in optimization theory to develop fast inference tools. In addition, we propose useful heuristic scores to identify the number of populations represented in a data set and a new hierarchical prior to detect weak population structure in the data. We test the variational algorithms on simulated data and illustrate using genotype data from the CEPH-Human Genome Diversity Panel. The variational algorithms are almost two orders of magnitude faster than STRUCTURE and achieve accuracies comparable to those of ADMIXTURE. Furthermore, our results show that the heuristic scores for choosing model complexity provide a reasonable range of values for the number of populations represented in the data, with minimal bias toward detecting structure when it is very weak. Our algorithm, fastSTRUCTURE, is freely available online at http://pritchardlab.stanford.edu/structure.html.
Genetic discovery for oil production and quality in sesame
DOI:10.1038/ncomms9609
URL
PMID:26477832
[本文引用: 2]
Oilseed crops are used to produce vegetable oil. Sesame (Sesamum indicum), an oilseed crop grown worldwide, has high oil content and a small diploid genome, but the genetic basis of oil production and quality is unclear. Here we sequence 705 diverse sesame varieties to construct a haplotype map of the sesame genome and de novo assemble two representative varieties to identify sequence variations. We investigate 56 agronomic traits in four environments and identify 549 associated loci. Examination of the major loci identifies 46 candidate causative genes, including genes related to oil content, fatty acid biosynthesis and yield. Several of the candidate genes for oil content encode enzymes involved in oil metabolism. Two major genes associated with lignification and black pigmentation in the seed coat are also associated with large variation in oil content. These findings may inform breeding and improvement strategies for a broad range of oilseed crops.
Haploview:Analysis and visualization of LD and haplotype maps
DOI:10.1093/bioinformatics/bth457
URL
PMID:15297300
[本文引用: 1]
UNLABELLED: Research over the last few years has revealed significant haplotype structure in the human genome. The characterization of these patterns, particularly in the context of medical genetic association studies, is becoming a routine research activity. Haploview is a software package that provides computation of linkage disequilibrium statistics and population haplotype patterns from primary genotype data in a visually appealing and interactive interface. AVAILABILITY: http://www.broad.mit.edu/mpg/haploview/ CONTACT: jcbarret@broad.mit.edu
ANNOVAR:functional annotation of genetic variants from high-throughput sequencing data
DOI:10.1093/nar/gkq603
URL
PMID:20601685
[本文引用: 2]
High-throughput sequencing platforms are generating massive amounts of genetic variation data for diverse genomes, but it remains a challenge to pinpoint a small subset of functionally important variants. To fill these unmet needs, we developed the ANNOVAR tool to annotate single nucleotide variants (SNVs) and insertions/deletions, such as examining their functional consequence on genes, inferring cytogenetic bands, reporting functional importance scores, finding variants in conserved regions, or identifying variants reported in the 1000 Genomes Project and dbSNP. ANNOVAR can utilize annotation databases from the UCSC Genome Browser or any annotation data set conforming to Generic Feature Format version 3 (GFF3). We also illustrate a 'variants reduction' protocol on 4.7 million SNVs and indels from a human genome, including two causal mutations for Miller syndrome, a rare recessive disease. Through a stepwise procedure, we excluded variants that are unlikely to be causal, and identified 20 candidate genes including the causal gene. Using a desktop computer, ANNOVAR requires approximately 4 min to perform gene-based annotation and approximately 15 min to perform variants reduction on 4.7 million variants, making it practical to handle hundreds of human genomes in a day. ANNOVAR is freely available at http://www.openbioinformatics.org/annovar/.
Gene expression regulation by upstream open reading frames and human disease
DOI:10.1371/journal.pgen.1003529
URL
PMID:23950723
[本文引用: 2]
Upstream open reading frames (uORFs) are major gene expression regulatory elements. In many eukaryotic mRNAs, one or more uORFs precede the initiation codon of the main coding region. Indeed, several studies have revealed that almost half of human transcripts present uORFs. Very interesting examples have shown that these uORFs can impact gene expression of the downstream main ORF by triggering mRNA decay or by regulating translation. Also, evidence from recent genetic and bioinformatic studies implicates disturbed uORF-mediated translational control in the etiology of many human diseases, including malignancies, metabolic or neurologic disorders, and inherited syndromes. In this review, we will briefly present the mechanisms through which uORFs regulate gene expression and how they can impact on the organism's response to different cell stress conditions. Then, we will emphasize the importance of these structures by illustrating, with specific examples, how disturbed uORF-mediated translational control can be involved in the etiology of human diseases, giving special importance to genotype-phenotype correlations. Identifying and studying more cases of uORF-altering mutations will help us to understand and establish genotype-phenotype associations, leading to advancements in diagnosis, prognosis, and treatment of many human disorders.
The Genome Analysis Toolkit:a MapReduce framework for analyzing next-generation DNA sequencing data
TOUSLED is a nuclear serine/threonine protein kinase that requires a coiled-coil region for oligomerization and catalytic activity
UBA1 and UBA2,two proteins that interact with UBP1,a multifunctional effector of pre-mRNA maturation in plants
KOBITO1 encodes a novel plasma membrane protein necessary for normal synthesis of cellulose during cell expansion in Arabidopsis
DOI:10.1105/tpc.002873
URL
PMID:12215501
[本文引用: 1]
The cell wall is the major limiting factor for plant growth. Wall extension is thought to result from the loosening of its structure. However, it is not known how this is coordinated with wall synthesis. We have identified two novel allelic cellulose-deficient dwarf mutants, kobito1-1 and kobito1-2 (kob1-1 and kob1-2). The cellulose deficiency was confirmed by the direct observation of microfibrils in most recent wall layers of elongating root cells. In contrast to the wild type, which showed transversely oriented parallel microfibrils, kob1 microfibrils were randomized and occluded by a layer of pectic material. No such changes were observed in another dwarf mutant, pom1, suggesting that the cellulose defect in kob1 is not an indirect result of the reduced cell elongation. Interestingly, in the meristematic zone of kob1 roots, microfibrils appeared unaltered compared with the wild type, suggesting a role for KOB1 preferentially in rapidly elongating cells. KOB1 was cloned and encodes a novel, highly conserved, plant-specific protein that is plasma membrane bound, as shown with a green fluorescent protein-KOB1 fusion protein. KOB1 mRNA was present in all organs investigated, and its overexpression did not cause visible phenotypic changes. KOB1 may be part of the cellulose synthesis machinery in elongating cells, or it may play a role in the coordination between cell elongation and cellulose synthesis.
Glycosyltransferase-like protein ABI8/ELD1/KOB1 promotes Arabidopsis hypocotyl elongation through regulating cellulose biosynthesis
Xyloglucan Xylosyltransferases XXT1,XXT2,and XXT5 and the Glucan Synthase CSLC4 form golgi-localized multiprotein complexes
DOI:10.1104/pp.112.199356
URL
PMID:22665445
[本文引用: 1]
Xyloglucan is the major hemicellulosic polysaccharide in the primary cell walls of most vascular dicotyledonous plants and has important structural and physiological functions in plant growth and development. In Arabidopsis (Arabidopsis thaliana), the 1,4-beta-glucan synthase, Cellulose Synthase-Like C4 (CSLC4), and three xylosyltransferases, XXT1, XXT2, and XXT5, act in the Golgi to form the xylosylated glucan backbone during xyloglucan biosynthesis. However, the functional organization of these enzymes in the Golgi membrane is currently unknown. In this study, we used bimolecular fluorescence complementation and in vitro pull-down assays to investigate the supramolecular organization of the CSLC4, XXT1, XXT2, and XXT5 proteins in Arabidopsis protoplasts. Quantification of bimolecular fluorescence complementation fluorescence by flow cytometry allowed us to perform competition assays that demonstrated the high probability of protein-protein complex formation in vivo and revealed differences in the abilities of these proteins to form multiprotein complexes. Results of in vitro pull-down assays using recombinant proteins confirmed that the physical interactions among XXTs occur through their catalytic domains. Additionally, coimmunoprecipitation of XXT2YFP and XXT5HA proteins from Arabidopsis protoplasts indicated that while the formation of the XXT2-XXT2 homocomplex involves disulfide bonds, the formation of the XXT2-XXT5 heterocomplex does not involve covalent interactions. The combined data allow us to propose that the proteins involved in xyloglucan biosynthesis function in a multiprotein complex composed of at least two homocomplexes, CSLC4-CSLC4 and XXT2-XXT2, and three heterocomplexes, XXT2-XXT5, XXT1-XXT2, and XXT5-CSLC4.
Mutation in xyloglucan 6-xylosytransferase results in abnormal root hair development in Oryza sativa
DOI:10.1093/jxb/eru189
URL
PMID:24834920
[本文引用: 1]
Root hairs are important for nutrient uptake, anchorage, and plant-microbe interactions. From a population of rice (Oryza sativa) mutagenized by ethyl methanesulfonate (EMS), a short root hair2 (srh2) mutant was identified. In hydroponic culture, srh2 seedlings were significantly reduced in root hair length. Bubble-like extrusions and irregular epidermal cells were observed at the tips of srh2 root hairs when grown under acidic conditions, suggesting the possible reduction of the tensile strength of the cell wall in this mutant. Map-based cloning identified a mutation in the gene encoding xyloglucan (XyG) 6-xylosyltransferase (OsXXT1). OsXXT1 displays more than 70% amino acid sequence identity with the previously characterized Arabidopsis thaliana XYG XYLOSYL TRANSFERASE 1 (AtXXT1) and XYG XYLOSYL TRANSFERASE 2 (AtXXT2), which catalyse the transfer of xylose onto beta-1,4-glucan chains. Furthermore, expression of the full-length coding sequence of OsXXT1 could complement the root hair defect, and slow growth and XyG synthesis in the Arabidopsis xxt1 xxt2 double mutant. Transgenic plants expressing the beta-glucuronidase (GUS) reporter under the control of the OsXXT1 promoter displayed GUS expression in multiple tissues, most prominently in root epidermal cells. These results demonstrate the importance of OsXXT1 in maintaining cell wall structure and tensile strength in rice, a typical grass species that contains relatively low XyG content in cell walls.
The role of plant cell wall polysaccharide composition in disease resistance
DOI:10.1016/j.tplants.2004.02.005
URL
[本文引用: 1]
AbstractThe high degree of structural complexity of plant cell wall polysaccharides has led to suggestions that some components might function as latent signal molecules that are released during pathogen infections and elicit defensive responses by the plant. However, there has been a paucity of genetic evidence supporting the idea that variation in cell wall composition plays a role in the outcome of host–pathogen interactions. Recently, several genetic studies have provided new lines of evidence implicating cell wall polysaccharides as factors in host–pathogen interactions.]]>
A gene encoding a chloroplast-targeted lipoxygenase in tomato leaves is transiently induced by wounding,systemin,and methyl jasmonate
DOI:10.1104/pp.114.3.1085
URL
PMID:9232884
[本文引用: 1]
We investigated the relationship between the expression of lipoxygenase (LOX) genes and the systemin-dependent wound response in tomato (Lycopersicon esculentum) leaves. A polymerase chain reaction-based approach was used to isolate two tomato Lox cDNAs, called TomLoxC and TomLoxD. Both TomLOXC and TomLOXD amino acid sequences possess an N-terminal extension of about 60 residues that were shown by in vitro uptake to function as transit peptides, targeting these proteins into the chloroplast. Within 30 to 50 min following wounding or systemin or methyl jasmonate treatments, the TomLoxD mRNA level increased and reached a maximum between 1 and 2 h. TomLoxC mRNA was not detectable in leaves and was not found following wounding, but it was found in ripening fruits, indicating that the two tomato Lox genes are regulated in different tissues by different processes. The results suggest that the TomLoxD gene is up-regulated in leaves in response to wounding and encodes a chloroplast LOX that may play a role as a component of the octadecanoid defense-signaling pathway.
昆虫对植物蛋白酶抑制素的诱导及适应机制
植物蛋白酶抑制素是植物重要的防御物质之一,一般是分子量较小的多肽或蛋白质,能够与昆虫消化道内的蛋白酶形成复合物,阻断或削弱蛋白酶对食物中蛋白的水解,使昆虫厌食或消化不良而致死。植物蛋白酶抑制素在植物体内一般是诱导表达的,昆虫取食危害后,导致某些植物在伤口产生一种寡聚糖信息素-蛋白酶抑制素诱导因子,蛋白酶抑制素诱导因子诱导叶片局部产生植物蛋白酶抑制素,并刺激产生信号物质系统肽,通过十八烷酸途径在一系列酶的作用下产生茉莉酸,茉莉酸与受体结合,活化植物蛋白酶抑制素基因。昆虫在长期取食植物蛋白酶抑制素后会在生理及行为上产生适应性而导致不敏感,适应方式主要包括:(1)改变肠道蛋白酶对蛋白酶抑制素的敏感性;(2) 水解蛋白酶抑制素;(3)过量取食及干扰产生蛋白酶抑制素的信号通道。由于昆虫能够对植物蛋白酶抑制素产生适应,因此合理利用植物蛋白酶抑制素的抗虫作用显得十分重要。
FtsH11 protease plays a critical role in Arabidopsis thermotolerance
DOI:10.1111/j.1365-313X.2006.02855.x
URL
PMID:16972866
[本文引用: 1]
Plants, as sessile organisms, employ multiple mechanisms to adapt to the seasonal and daily temperature fluctuations associated with their habitats. Here, we provide genetic and physiological evidence that the FtsH11 protease of Arabidopsis contributes to the overall tolerance of the plant to elevated temperatures. To identify the various mechanisms of thermotolerance in plants, we isolated a series of Arabidopsis thaliana thermo-sensitive mutants (atts) that fail to acquire thermotolerance after pre-conditioning at 38 degrees C. Two allelic mutants, atts244 and atts405, were found to be both highly susceptible to moderately elevated temperatures and defective in acquired thermotolerance. The growth and development of the mutant plants at all stages examined were arrested after exposure to temperatures above 30 degrees C, which are permissive conditions for wild-type plants. The affected gene in atts244 was identified through map-based cloning and encodes a chloroplast targeted FtsH protease, FtsH11. The Arabidopsis genome contains 12 predicted FtsH protease genes, with all previously characterized FtsH genes playing roles in the alleviation of light stress through the degradation of unassembled thylakoid membrane proteins and photodamaged photosystem II D1 protein. Photosynthetic capability, as measured by chlorophyll content (chl a/b ratios) and PSII quantum yield, is greatly reduced in the leaves of FtsH11 mutants when exposed to the moderately high temperature of 30 degrees C. Under high light conditions, however, FtsH11 mutants and wild-type plants showed no significant difference in photosynthesis capacity. Our results support a direct role for the A. thaliana FtsH11-encoded protease in thermotolerance, a function previously reported for bacterial and yeast FtsH proteases but not for those from plants.
Cloning and use of the ms9 gene from maize:US20160024520A1

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}