农作物遗传多样性农家保护的现状及前景
1
2002
... 种质资源创新对农作物可持续发展起到决定性的作用,人为定向选择导致种质资源遗传多样性逐渐减少,耐逆性和稳定性减弱,再加上生态环境日趋恶化,农作物生产面临严峻的考验[1].种质资源是作物遗传改良和相关基础研究的物质基础,积累了大量自然和人为的遗传变异,保证了作物遗传基因的丰富性[2].因此,在遗传学研究上,积累丰富遗传多样性的种质资源可以发挥其多方面的潜力优势. ...
农作物多基因型种群育种及种子生产技术体系
1
2012
... 种质资源创新对农作物可持续发展起到决定性的作用,人为定向选择导致种质资源遗传多样性逐渐减少,耐逆性和稳定性减弱,再加上生态环境日趋恶化,农作物生产面临严峻的考验[1].种质资源是作物遗传改良和相关基础研究的物质基础,积累了大量自然和人为的遗传变异,保证了作物遗传基因的丰富性[2].因此,在遗传学研究上,积累丰富遗传多样性的种质资源可以发挥其多方面的潜力优势. ...
甘蓝型油菜MAGIC群体的遗传结构分析及应用
2
2015
... 作物的重要农艺性状大多是受多基因控制的数量性状,而耐逆性大多属于复杂的数量性状.通过遗传定位可发掘到控制数量性状的位点(quantitative trait locus,QTL),其中构建作图群体是决定数量性状定位准确性的关键因素[3].目前,作物中用于遗传定位的群体主要可以分为3类:传统双亲作图群体、自然群体及新一代作图群体(图1).传统双亲作图群体主要包括F2群体、DH(doubled haploid)群体、回交(backcross,BC)群体和重组自交系(recombinant inbred lines,RIL)群体.传统双亲作图群体的局限性是遗传多样性不足、QTL定位准确性较低和推广应用时间较长等[3].自然群体用于遗传研究还属于较新的研究范围,Thornsberry等[4]首次将关联分析方法应用于植物遗传研究领域,目前仍广泛应用于数量性状的定位分析.随着数量遗传学的发展,促进了作物的新一代作图群体的产生与应用.以巢式关联图谱(nested association mapping,NAM)和多亲本高世代互交(multi-parent advanced qeneration intercross,MAGIC)群体为代表的新一代作图群体是通过多亲本杂交后自交产生,经过多次减数分裂后,它们包含更多的重组事件,可以构建更大的群体以及丰富的遗传多样性. ...
... [3].自然群体用于遗传研究还属于较新的研究范围,Thornsberry等[4]首次将关联分析方法应用于植物遗传研究领域,目前仍广泛应用于数量性状的定位分析.随着数量遗传学的发展,促进了作物的新一代作图群体的产生与应用.以巢式关联图谱(nested association mapping,NAM)和多亲本高世代互交(multi-parent advanced qeneration intercross,MAGIC)群体为代表的新一代作图群体是通过多亲本杂交后自交产生,经过多次减数分裂后,它们包含更多的重组事件,可以构建更大的群体以及丰富的遗传多样性. ...
Dwarf8 polymorphisms associate with variation in flowering time
1
2001
... 作物的重要农艺性状大多是受多基因控制的数量性状,而耐逆性大多属于复杂的数量性状.通过遗传定位可发掘到控制数量性状的位点(quantitative trait locus,QTL),其中构建作图群体是决定数量性状定位准确性的关键因素[3].目前,作物中用于遗传定位的群体主要可以分为3类:传统双亲作图群体、自然群体及新一代作图群体(图1).传统双亲作图群体主要包括F2群体、DH(doubled haploid)群体、回交(backcross,BC)群体和重组自交系(recombinant inbred lines,RIL)群体.传统双亲作图群体的局限性是遗传多样性不足、QTL定位准确性较低和推广应用时间较长等[3].自然群体用于遗传研究还属于较新的研究范围,Thornsberry等[4]首次将关联分析方法应用于植物遗传研究领域,目前仍广泛应用于数量性状的定位分析.随着数量遗传学的发展,促进了作物的新一代作图群体的产生与应用.以巢式关联图谱(nested association mapping,NAM)和多亲本高世代互交(multi-parent advanced qeneration intercross,MAGIC)群体为代表的新一代作图群体是通过多亲本杂交后自交产生,经过多次减数分裂后,它们包含更多的重组事件,可以构建更大的群体以及丰富的遗传多样性. ...
Natural variation for seed dormancy in Arabidopsis is regulated by additive genetic and molecular pathways
1
2010
... MAGIC群体更容易获得多个优良NAM群体是按照星型杂交方案将1个中心亲本与多个亲本杂交构建得来的巢式群体[5⇓-7].有研究团队[8⇓-10]采用单粒传法(single seed descent,SSD)将25个玉米自交系与B73杂交,构建了包含200个RIL的NAM群体.但NAM群体只有1个共同的亲本,无法研究剩余亲本间QTL的相互作用[11].因此,有研究[12-13]提出了用MAGIC解决目前已有作图群体中所存在的问题,并指出MAGIC群体同时适用于连锁分析和关联分析这2种方法进行QTL定位,并且在构建过程中实现了多亲本的聚合杂交、重组产生的变异多样性、LD衰减更快等目的.在育种上,基因聚合的材料,并聚合了多个亲本的遗传背景,对其育种价值和遗传价值趋近于理想群体[14].因此,此类群体在作物育种应用有着重要的意义. ...
A genome-wide meta-analysis of rice blast resistance genes and quantitative trait loci provides new insights into partial and complete resistance
1
2008
... MAGIC群体更容易获得多个优良NAM群体是按照星型杂交方案将1个中心亲本与多个亲本杂交构建得来的巢式群体[5⇓-7].有研究团队[8⇓-10]采用单粒传法(single seed descent,SSD)将25个玉米自交系与B73杂交,构建了包含200个RIL的NAM群体.但NAM群体只有1个共同的亲本,无法研究剩余亲本间QTL的相互作用[11].因此,有研究[12-13]提出了用MAGIC解决目前已有作图群体中所存在的问题,并指出MAGIC群体同时适用于连锁分析和关联分析这2种方法进行QTL定位,并且在构建过程中实现了多亲本的聚合杂交、重组产生的变异多样性、LD衰减更快等目的.在育种上,基因聚合的材料,并聚合了多个亲本的遗传背景,对其育种价值和遗传价值趋近于理想群体[14].因此,此类群体在作物育种应用有着重要的意义. ...
Genetic design and statistical power of nested association mapping in maize
1
2008
... MAGIC群体更容易获得多个优良NAM群体是按照星型杂交方案将1个中心亲本与多个亲本杂交构建得来的巢式群体[5⇓-7].有研究团队[8⇓-10]采用单粒传法(single seed descent,SSD)将25个玉米自交系与B73杂交,构建了包含200个RIL的NAM群体.但NAM群体只有1个共同的亲本,无法研究剩余亲本间QTL的相互作用[11].因此,有研究[12-13]提出了用MAGIC解决目前已有作图群体中所存在的问题,并指出MAGIC群体同时适用于连锁分析和关联分析这2种方法进行QTL定位,并且在构建过程中实现了多亲本的聚合杂交、重组产生的变异多样性、LD衰减更快等目的.在育种上,基因聚合的材料,并聚合了多个亲本的遗传背景,对其育种价值和遗传价值趋近于理想群体[14].因此,此类群体在作物育种应用有着重要的意义. ...
Structure of linkage disequilibrium and phenotypic associations in the maize genome
1
2001
... MAGIC群体更容易获得多个优良NAM群体是按照星型杂交方案将1个中心亲本与多个亲本杂交构建得来的巢式群体[5⇓-7].有研究团队[8⇓-10]采用单粒传法(single seed descent,SSD)将25个玉米自交系与B73杂交,构建了包含200个RIL的NAM群体.但NAM群体只有1个共同的亲本,无法研究剩余亲本间QTL的相互作用[11].因此,有研究[12-13]提出了用MAGIC解决目前已有作图群体中所存在的问题,并指出MAGIC群体同时适用于连锁分析和关联分析这2种方法进行QTL定位,并且在构建过程中实现了多亲本的聚合杂交、重组产生的变异多样性、LD衰减更快等目的.在育种上,基因聚合的材料,并聚合了多个亲本的遗传背景,对其育种价值和遗传价值趋近于理想群体[14].因此,此类群体在作物育种应用有着重要的意义. ...
Multi-parent advanced generation inter-cross (MAGIC) populations in rice:progress and potential for genetics research and breeding
1
2013
... MAGIC群体更容易获得多个优良NAM群体是按照星型杂交方案将1个中心亲本与多个亲本杂交构建得来的巢式群体[5⇓-7].有研究团队[8⇓-10]采用单粒传法(single seed descent,SSD)将25个玉米自交系与B73杂交,构建了包含200个RIL的NAM群体.但NAM群体只有1个共同的亲本,无法研究剩余亲本间QTL的相互作用[11].因此,有研究[12-13]提出了用MAGIC解决目前已有作图群体中所存在的问题,并指出MAGIC群体同时适用于连锁分析和关联分析这2种方法进行QTL定位,并且在构建过程中实现了多亲本的聚合杂交、重组产生的变异多样性、LD衰减更快等目的.在育种上,基因聚合的材料,并聚合了多个亲本的遗传背景,对其育种价值和遗传价值趋近于理想群体[14].因此,此类群体在作物育种应用有着重要的意义. ...
High-resolution molecular karyotyping uncovers pairing between ancestrally related Brassica chromosomes
1
2014
... MAGIC群体更容易获得多个优良NAM群体是按照星型杂交方案将1个中心亲本与多个亲本杂交构建得来的巢式群体[5⇓-7].有研究团队[8⇓-10]采用单粒传法(single seed descent,SSD)将25个玉米自交系与B73杂交,构建了包含200个RIL的NAM群体.但NAM群体只有1个共同的亲本,无法研究剩余亲本间QTL的相互作用[11].因此,有研究[12-13]提出了用MAGIC解决目前已有作图群体中所存在的问题,并指出MAGIC群体同时适用于连锁分析和关联分析这2种方法进行QTL定位,并且在构建过程中实现了多亲本的聚合杂交、重组产生的变异多样性、LD衰减更快等目的.在育种上,基因聚合的材料,并聚合了多个亲本的遗传背景,对其育种价值和遗传价值趋近于理想群体[14].因此,此类群体在作物育种应用有着重要的意义. ...
Multiparent intercross populations in analysis of quantitative traits
1
2012
... MAGIC群体更容易获得多个优良NAM群体是按照星型杂交方案将1个中心亲本与多个亲本杂交构建得来的巢式群体[5⇓-7].有研究团队[8⇓-10]采用单粒传法(single seed descent,SSD)将25个玉米自交系与B73杂交,构建了包含200个RIL的NAM群体.但NAM群体只有1个共同的亲本,无法研究剩余亲本间QTL的相互作用[11].因此,有研究[12-13]提出了用MAGIC解决目前已有作图群体中所存在的问题,并指出MAGIC群体同时适用于连锁分析和关联分析这2种方法进行QTL定位,并且在构建过程中实现了多亲本的聚合杂交、重组产生的变异多样性、LD衰减更快等目的.在育种上,基因聚合的材料,并聚合了多个亲本的遗传背景,对其育种价值和遗传价值趋近于理想群体[14].因此,此类群体在作物育种应用有着重要的意义. ...
From mutations to MAGIC:resources for gene discovery,validation and delivery in crop plants
2
2007
... MAGIC群体更容易获得多个优良NAM群体是按照星型杂交方案将1个中心亲本与多个亲本杂交构建得来的巢式群体[5⇓-7].有研究团队[8⇓-10]采用单粒传法(single seed descent,SSD)将25个玉米自交系与B73杂交,构建了包含200个RIL的NAM群体.但NAM群体只有1个共同的亲本,无法研究剩余亲本间QTL的相互作用[11].因此,有研究[12-13]提出了用MAGIC解决目前已有作图群体中所存在的问题,并指出MAGIC群体同时适用于连锁分析和关联分析这2种方法进行QTL定位,并且在构建过程中实现了多亲本的聚合杂交、重组产生的变异多样性、LD衰减更快等目的.在育种上,基因聚合的材料,并聚合了多个亲本的遗传背景,对其育种价值和遗传价值趋近于理想群体[14].因此,此类群体在作物育种应用有着重要的意义. ...
... Cavanagh等[12]向植物界引入MAGIC群体,该群体构建过程主要包括3个阶段:亲本选择阶段、互交重组阶段和子代纯合阶段.构建1个MAGIC群体要求所有亲本的遗传因子都要融合到后代中,2n个亲本互交n代才能实现融合,再加上重组和纯合至少需要10世代以上.它的一个重要特征是由多亲本互交后自交而得来的,包含多个亲本的遗传组成,不会出现后代衰退和多态性下降的现象.因此在作物遗传研究中,其最大的优点是可以建立一个包含很多株系的群体,这些株系拥有丰富的遗传多样性基因池. ...
Genome-wide comparative diversity uncovers multiple targets of selection for inprovement in hexaploid wheat landraces and cultivars
2
2013
... MAGIC群体更容易获得多个优良NAM群体是按照星型杂交方案将1个中心亲本与多个亲本杂交构建得来的巢式群体[5⇓-7].有研究团队[8⇓-10]采用单粒传法(single seed descent,SSD)将25个玉米自交系与B73杂交,构建了包含200个RIL的NAM群体.但NAM群体只有1个共同的亲本,无法研究剩余亲本间QTL的相互作用[11].因此,有研究[12-13]提出了用MAGIC解决目前已有作图群体中所存在的问题,并指出MAGIC群体同时适用于连锁分析和关联分析这2种方法进行QTL定位,并且在构建过程中实现了多亲本的聚合杂交、重组产生的变异多样性、LD衰减更快等目的.在育种上,基因聚合的材料,并聚合了多个亲本的遗传背景,对其育种价值和遗传价值趋近于理想群体[14].因此,此类群体在作物育种应用有着重要的意义. ...
... Application of MAGIC group in plant genetics research
Table 1 作物
Crop | 类型
Type | 亲本选择
Parental selection | 杂交方式
Hybridization | 群体规模
Group size | 基因型鉴定
Genotyping | 研究性状
Research trait | 分析方法
Analytical method | 参考文献
Reference |
拟南芥
Arabidopsis | –
| 19亲本
| 随机杂交
| 1026 F4
| 1260 SNPs
| 抽薹期
| QTL
| [26]
|
| 水稻Rice | 籼稻 | 4亲本DC1 | 双列杂交 | 495 F6 | 6K SNP芯片 | 籽粒产量性状、穗相关性状、分蘖相关性状、抽穗期、苗期和株高
| GWAS | [23,28-31] |
| | 4亲本DC2 | 双列杂交 | 525 F6 | | | |
| | 8亲本
| Funnel杂交
| 668 F6
| 55K SNP芯片,
GBS | | |
| 籼稻 | 8亲本 | Funnel杂交 | 1328 S7 | GBS: 17387 SNP | 产量性状、稻米品质、开花期、株高、粒型、抗旱性、耐涝性、耐盐性、抗病性(稻瘟病、白叶枯病、褐斑病)
| QTL,GWAS | [32⇓-34] |
| | MAGIC PLUS | ― | 144 S8 | | | |
| | MAGIC PLUS
DH系 | ―
| 76 DH
| | | |
| 粳稻 | 8亲本 | Funnel杂交 | 500 S5 | | | |
| 籼―粳 | GLOBAL MAGIC | ― | 1402 S7 | | | |
小麦Wheat
| 春小麦
| 4亲本
| Funnel杂交
| 1579 F6
| 826 DArTs、283SNPs、53SSRs,9K SNP芯片,90K SNP芯片 | 株高、籽粒重、
胚芽鞘长度
| LMC,QTL
| [13,35-36]
|
| 冬小麦
| 8亲本
| Funnel杂交
| 1091 F7
| 90K SNP芯片
| 产量、开花期、
病害和株高 | LMC,QTL
| [37-38]
|
| 全麦粉
| 4亲本
| Funnel杂交
| 332 F6
| SSR
| 产量性状、
抗白粉病 | QTL,LMC
| [39]
|
| 欧洲
小麦 | 60亲本
| 随机杂交
| 1000 S4
| 9K SNP芯片
| 抽穗期
| LDA,GWAS
| [40]
|
| | | | | 14个KASPar SNP | | | |
大麦
Hordeum | – | 8亲本 | Funnel杂交 | 5000 DH | 9K SNP芯片 | 开花期 | QTL | [41] |
| 8亲本 | Funnel杂交 | 642 F5 | 9K SNP芯片 | 黄斑病 | LMC,QTL | [42] |
| 棉花Cotton | 陆地棉 | 11亲本 | 随机杂交 | 547 F5 | GBS | 纤维品质 | GWAS,QTL | [43] |
| | 8亲本
| 随机杂交
| 960 MLs
| SLAF-seq
| 开花期、株型、
果枝数 | GWAS,QTL
| [44]
|
| | 11亲本 | 随机杂交 | 550 RIL | 47K SNP芯片 | 株高、茎秆质量 | GWAS,QTL | [45] |
高粱
Sorghum | –
| 29亲本
| 随机杂交
| 1000 F6
| GBS
| 株高
| GWAS,QTL
| [46]
|
作物
Crop | 类型
Type | 亲本选择
Parental selection | 杂交方式
Hybridization | 群体规模
Group size | 基因型鉴定
Genotyping | 研究性状
Research trait | 分析方法
Analytical method | 参考文献
Reference |
烟草
Tobacco | –
| 8亲本
| 双列杂交
| 600 F4
| 430K 芯片
| 株型、烟叶品质
| GWAS
| [47]
|
玉米
Maize | –
| 8亲本
| Funnel杂交
| 1636 F6
| GBS
| 开花期、株高、
穗位高 | LMC,AM,QTL | [48]
|
| | 4亲本
| Funnel杂交
| 1149 S3
| 118K SNP 芯片
| 株高、穗高、散
粉期、吐丝期 | QTL,GWAS
| [49]
|
| | 8亲本
| Funnel杂交
| 401 RIL
| 56K SNP 芯片
| 苗重、苗长和
黄萎病 | QTL,NAM
| [50]
|
| | 8亲本
| Funnel杂交
| 700F6
| 110K SNP
芯片 | 镰孢菌穗腐病
| LMC,AM,QTL | [51]
|
| | 8亲本
| Funnel杂交
| 406 RIL
| 100K SNP
芯片,GBS | 发芽率、叶绿素
和出苗期 | GWAS,QTL
| [52]
|
| | 8亲本
| Funnel杂交
| 368 MLs
| 1000K SNP芯片,GBS | 籽粒含油量
| GWAS,QTL
| [53]
|
| | 16亲本
| Funnel杂交
| 513 RIL
| 500K SNP芯片
| 株高、叶形态、
开花期、穗大小 | GWAS
| [54]
|
| | 4亲本
| Funnel杂交
| 1044 F6
| 50K SNP芯片
| 产量、吐丝期、
株高 | QTL,AM
| [55]
|
油菜
Brassica napus | 甘蓝型
| 8亲本
| 随机杂交
| 680 F6
| –
| 始花期、抗病性
| SSD
| [56]
|
Fn表示构建F1的杂交过程和F1自交过程的总代数,Sn表示F1植株自交的代数,DH(doubled haploid)表示双单倍体系,GBS(genotyping by sequencing)表示基于测序的基因分型,QTL(quantitative trait locus)表示QTL定位,LDA(linkage disequilibrium analysis)表示连锁不平衡分析,LMC(linkage map construction)表示连锁图谱构建,AM(association mapping)表示关联作图,GWAS(genome wide association study)表示全基因组关联分析,SNP(single nucleotide polymorphisms)表示单核苷酸多态性,Funnel杂交表示“漏斗”型杂交,SSR(simple sequence repeat)表示简单重复序列,SSD(single seed descent)表示单粒传法,RIL(recombinant inbred lines)表示重组自交系,ML(lines)表示自交系,NAM(nested association map)表示巢式关联图谱 ...
Analysis of natural allelic variation in Arabidopsis using a multiparent recombinant inbred line population
1
2011
... MAGIC群体更容易获得多个优良NAM群体是按照星型杂交方案将1个中心亲本与多个亲本杂交构建得来的巢式群体[5⇓-7].有研究团队[8⇓-10]采用单粒传法(single seed descent,SSD)将25个玉米自交系与B73杂交,构建了包含200个RIL的NAM群体.但NAM群体只有1个共同的亲本,无法研究剩余亲本间QTL的相互作用[11].因此,有研究[12-13]提出了用MAGIC解决目前已有作图群体中所存在的问题,并指出MAGIC群体同时适用于连锁分析和关联分析这2种方法进行QTL定位,并且在构建过程中实现了多亲本的聚合杂交、重组产生的变异多样性、LD衰减更快等目的.在育种上,基因聚合的材料,并聚合了多个亲本的遗传背景,对其育种价值和遗传价值趋近于理想群体[14].因此,此类群体在作物育种应用有着重要的意义. ...
The Collaborative Cross,a community resource for the genetic analysis of complex traits
2
2004
... Churchill等[15]以动物及人类复杂性状遗传基础为研究目标,成立了1个复杂性状协作组(complex trait consortium,CTC).该组织机构在2006年概述了一种称为联合杂交(colla-borative cross,CC)的策略,该策略是从一组遗传多样化的近交系小鼠品系构建大量的重组近交系集合,并且这种策略已经成功地用于小鼠和果蝇中QTL的定位[15⇓⇓⇓⇓⇓-21]. ...
... [15⇓⇓⇓⇓⇓-21]. ...
Genome-wide association studies of 14 agronomic traits in rice landraces
1
2010
... Churchill等[15]以动物及人类复杂性状遗传基础为研究目标,成立了1个复杂性状协作组(complex trait consortium,CTC).该组织机构在2006年概述了一种称为联合杂交(colla-borative cross,CC)的策略,该策略是从一组遗传多样化的近交系小鼠品系构建大量的重组近交系集合,并且这种策略已经成功地用于小鼠和果蝇中QTL的定位[15⇓⇓⇓⇓⇓-21]. ...
A method for fine mapping quantitative trait loci in outbredanimal stocks
1
2000
... Churchill等[15]以动物及人类复杂性状遗传基础为研究目标,成立了1个复杂性状协作组(complex trait consortium,CTC).该组织机构在2006年概述了一种称为联合杂交(colla-borative cross,CC)的策略,该策略是从一组遗传多样化的近交系小鼠品系构建大量的重组近交系集合,并且这种策略已经成功地用于小鼠和果蝇中QTL的定位[15⇓⇓⇓⇓⇓-21]. ...
High-resolution genetic mapping in the diversity outbred mouse population identifies Apobec1 as a candidate gene for atherosclerosis
1
2014
... Churchill等[15]以动物及人类复杂性状遗传基础为研究目标,成立了1个复杂性状协作组(complex trait consortium,CTC).该组织机构在2006年概述了一种称为联合杂交(colla-borative cross,CC)的策略,该策略是从一组遗传多样化的近交系小鼠品系构建大量的重组近交系集合,并且这种策略已经成功地用于小鼠和果蝇中QTL的定位[15⇓⇓⇓⇓⇓-21]. ...
Genetic dissection of a model complex trait using the Drosophila Synthetic population resource
1
2012
... Churchill等[15]以动物及人类复杂性状遗传基础为研究目标,成立了1个复杂性状协作组(complex trait consortium,CTC).该组织机构在2006年概述了一种称为联合杂交(colla-borative cross,CC)的策略,该策略是从一组遗传多样化的近交系小鼠品系构建大量的重组近交系集合,并且这种策略已经成功地用于小鼠和果蝇中QTL的定位[15⇓⇓⇓⇓⇓-21]. ...
Joint estimates of quantitative trait locus effect and frequency using synthetic recombinant populations of Drosophila melanogaster
1
2007
... Churchill等[15]以动物及人类复杂性状遗传基础为研究目标,成立了1个复杂性状协作组(complex trait consortium,CTC).该组织机构在2006年概述了一种称为联合杂交(colla-borative cross,CC)的策略,该策略是从一组遗传多样化的近交系小鼠品系构建大量的重组近交系集合,并且这种策略已经成功地用于小鼠和果蝇中QTL的定位[15⇓⇓⇓⇓⇓-21]. ...
Fine-mapping nicotine resistance loci in Drosophila using a multiparent advanced generation inter-cross population
1
2014
... Churchill等[15]以动物及人类复杂性状遗传基础为研究目标,成立了1个复杂性状协作组(complex trait consortium,CTC).该组织机构在2006年概述了一种称为联合杂交(colla-borative cross,CC)的策略,该策略是从一组遗传多样化的近交系小鼠品系构建大量的重组近交系集合,并且这种策略已经成功地用于小鼠和果蝇中QTL的定位[15⇓⇓⇓⇓⇓-21]. ...
水稻MAGIC群体的分子基础及生态响应的特性
1
2012
... MAGIC群体既克服了双亲本杂交群体遗传基础狭窄的限制,又弥补了自然群体进行关联分析的不足,逐渐成为一种替代型的作图群体[22].MAGIC群体在作物上有着自己独特的优势,可以同时定位多个主效基因和QTL,发挥其群体多样性高的优势,并且探讨包含来自不同亲本的多个等位基因及其对某个性状的影响;可以同时评估多个QTL在不同遗传背景下的表现,从而可以间接反映出不同遗传背景的作用;可以用于定位作图,选择优异材料的育种性状作为亲本可以产生大量的遗传变异,直接将优良个体用作育种材料,以达到育种群体和定位群体的相互融合,定位到的QTL可以直接指导育种[23-24]. ...
QTL mapping for agronomic traits using Multi-parent Advanced Generation Inter-Cross (MAGIC) populations derived from diverse elite Indica rice lines
3
2016
... MAGIC群体既克服了双亲本杂交群体遗传基础狭窄的限制,又弥补了自然群体进行关联分析的不足,逐渐成为一种替代型的作图群体[22].MAGIC群体在作物上有着自己独特的优势,可以同时定位多个主效基因和QTL,发挥其群体多样性高的优势,并且探讨包含来自不同亲本的多个等位基因及其对某个性状的影响;可以同时评估多个QTL在不同遗传背景下的表现,从而可以间接反映出不同遗传背景的作用;可以用于定位作图,选择优异材料的育种性状作为亲本可以产生大量的遗传变异,直接将优良个体用作育种材料,以达到育种群体和定位群体的相互融合,定位到的QTL可以直接指导育种[23-24]. ...
... Application of MAGIC group in plant genetics research
Table 1 作物
Crop | 类型
Type | 亲本选择
Parental selection | 杂交方式
Hybridization | 群体规模
Group size | 基因型鉴定
Genotyping | 研究性状
Research trait | 分析方法
Analytical method | 参考文献
Reference |
拟南芥
Arabidopsis | –
| 19亲本
| 随机杂交
| 1026 F4
| 1260 SNPs
| 抽薹期
| QTL
| [26]
|
| 水稻Rice | 籼稻 | 4亲本DC1 | 双列杂交 | 495 F6 | 6K SNP芯片 | 籽粒产量性状、穗相关性状、分蘖相关性状、抽穗期、苗期和株高
| GWAS | [23,28-31] |
| | 4亲本DC2 | 双列杂交 | 525 F6 | | | |
| | 8亲本
| Funnel杂交
| 668 F6
| 55K SNP芯片,
GBS | | |
| 籼稻 | 8亲本 | Funnel杂交 | 1328 S7 | GBS: 17387 SNP | 产量性状、稻米品质、开花期、株高、粒型、抗旱性、耐涝性、耐盐性、抗病性(稻瘟病、白叶枯病、褐斑病)
| QTL,GWAS | [32⇓-34] |
| | MAGIC PLUS | ― | 144 S8 | | | |
| | MAGIC PLUS
DH系 | ―
| 76 DH
| | | |
| 粳稻 | 8亲本 | Funnel杂交 | 500 S5 | | | |
| 籼―粳 | GLOBAL MAGIC | ― | 1402 S7 | | | |
小麦Wheat
| 春小麦
| 4亲本
| Funnel杂交
| 1579 F6
| 826 DArTs、283SNPs、53SSRs,9K SNP芯片,90K SNP芯片 | 株高、籽粒重、
胚芽鞘长度
| LMC,QTL
| [13,35-36]
|
| 冬小麦
| 8亲本
| Funnel杂交
| 1091 F7
| 90K SNP芯片
| 产量、开花期、
病害和株高 | LMC,QTL
| [37-38]
|
| 全麦粉
| 4亲本
| Funnel杂交
| 332 F6
| SSR
| 产量性状、
抗白粉病 | QTL,LMC
| [39]
|
| 欧洲
小麦 | 60亲本
| 随机杂交
| 1000 S4
| 9K SNP芯片
| 抽穗期
| LDA,GWAS
| [40]
|
| | | | | 14个KASPar SNP | | | |
大麦
Hordeum | – | 8亲本 | Funnel杂交 | 5000 DH | 9K SNP芯片 | 开花期 | QTL | [41] |
| 8亲本 | Funnel杂交 | 642 F5 | 9K SNP芯片 | 黄斑病 | LMC,QTL | [42] |
| 棉花Cotton | 陆地棉 | 11亲本 | 随机杂交 | 547 F5 | GBS | 纤维品质 | GWAS,QTL | [43] |
| | 8亲本
| 随机杂交
| 960 MLs
| SLAF-seq
| 开花期、株型、
果枝数 | GWAS,QTL
| [44]
|
| | 11亲本 | 随机杂交 | 550 RIL | 47K SNP芯片 | 株高、茎秆质量 | GWAS,QTL | [45] |
高粱
Sorghum | –
| 29亲本
| 随机杂交
| 1000 F6
| GBS
| 株高
| GWAS,QTL
| [46]
|
作物
Crop | 类型
Type | 亲本选择
Parental selection | 杂交方式
Hybridization | 群体规模
Group size | 基因型鉴定
Genotyping | 研究性状
Research trait | 分析方法
Analytical method | 参考文献
Reference |
烟草
Tobacco | –
| 8亲本
| 双列杂交
| 600 F4
| 430K 芯片
| 株型、烟叶品质
| GWAS
| [47]
|
玉米
Maize | –
| 8亲本
| Funnel杂交
| 1636 F6
| GBS
| 开花期、株高、
穗位高 | LMC,AM,QTL | [48]
|
| | 4亲本
| Funnel杂交
| 1149 S3
| 118K SNP 芯片
| 株高、穗高、散
粉期、吐丝期 | QTL,GWAS
| [49]
|
| | 8亲本
| Funnel杂交
| 401 RIL
| 56K SNP 芯片
| 苗重、苗长和
黄萎病 | QTL,NAM
| [50]
|
| | 8亲本
| Funnel杂交
| 700F6
| 110K SNP
芯片 | 镰孢菌穗腐病
| LMC,AM,QTL | [51]
|
| | 8亲本
| Funnel杂交
| 406 RIL
| 100K SNP
芯片,GBS | 发芽率、叶绿素
和出苗期 | GWAS,QTL
| [52]
|
| | 8亲本
| Funnel杂交
| 368 MLs
| 1000K SNP芯片,GBS | 籽粒含油量
| GWAS,QTL
| [53]
|
| | 16亲本
| Funnel杂交
| 513 RIL
| 500K SNP芯片
| 株高、叶形态、
开花期、穗大小 | GWAS
| [54]
|
| | 4亲本
| Funnel杂交
| 1044 F6
| 50K SNP芯片
| 产量、吐丝期、
株高 | QTL,AM
| [55]
|
油菜
Brassica napus | 甘蓝型
| 8亲本
| 随机杂交
| 680 F6
| –
| 始花期、抗病性
| SSD
| [56]
|
Fn表示构建F1的杂交过程和F1自交过程的总代数,Sn表示F1植株自交的代数,DH(doubled haploid)表示双单倍体系,GBS(genotyping by sequencing)表示基于测序的基因分型,QTL(quantitative trait locus)表示QTL定位,LDA(linkage disequilibrium analysis)表示连锁不平衡分析,LMC(linkage map construction)表示连锁图谱构建,AM(association mapping)表示关联作图,GWAS(genome wide association study)表示全基因组关联分析,SNP(single nucleotide polymorphisms)表示单核苷酸多态性,Funnel杂交表示“漏斗”型杂交,SSR(simple sequence repeat)表示简单重复序列,SSD(single seed descent)表示单粒传法,RIL(recombinant inbred lines)表示重组自交系,ML(lines)表示自交系,NAM(nested association map)表示巢式关联图谱 ...
... Meng等[28]构建了5个水稻MAGIC群体,并利用这5个水稻MAGIC群体进行了水稻苗期对铁、锌和铝毒害响应的关联分析,分别检测到21个与铁、铝胁迫相关的QTL和30个与锌胁迫相关的QTL.此外,Meng等[23,29]同时利用这些群体对水稻株高、产量、产量构成因素和抽穗期等14个农艺性状进行全基因组关联分析,得到除第7、9和11号染色体以外的9条染色体上共检测到26个显著的QTL定位.Ponce等[30]分别利用2个4亲本和8亲本的籼稻MAGIC群体,使用55K SNP和GBS(genotyping by sequencing)测序分别进行了基因分型,鉴定与粒长、粒宽、粒长宽比、粒厚和千粒重相关的QTL和SNP.结果显示,5个与粒级相关的性状共鉴定出18个QTL,解释了6.43%~63.35%的总表型方差.其中12个QTL与克隆基因GS3、GW5/qSW5、GW7/GL7/SLG7和GW8/OsSPL16定位一致,前2个基因对粒长和粒宽的影响最大.此外,MAGIC群体也被用于对水稻单倍体等位基因挖掘、稻米品质的QTL定位分析和生物强化等研究中. ...
Genotype Probabilities at intermediate generations in the construction of recombinant inbred lines
1
2012
... MAGIC群体既克服了双亲本杂交群体遗传基础狭窄的限制,又弥补了自然群体进行关联分析的不足,逐渐成为一种替代型的作图群体[22].MAGIC群体在作物上有着自己独特的优势,可以同时定位多个主效基因和QTL,发挥其群体多样性高的优势,并且探讨包含来自不同亲本的多个等位基因及其对某个性状的影响;可以同时评估多个QTL在不同遗传背景下的表现,从而可以间接反映出不同遗传背景的作用;可以用于定位作图,选择优异材料的育种性状作为亲本可以产生大量的遗传变异,直接将优良个体用作育种材料,以达到育种群体和定位群体的相互融合,定位到的QTL可以直接指导育种[23-24]. ...
利用水稻4亲本MAGIC群体进行粒形和株型的遗传分析
1
2017
... 目前,MAGIC群体主要基于以下2点进行遗传定位,一是利用基因型和表型数据进行关联分析,二是基于标记构建单倍型的连锁分析[25].关联分析在MAGIC群体中一般进行重要性状的遗传定位.与连锁定位相比,关联分析广度大,主要基于群体中普遍存在连锁不平衡现象,因此可以在同一座位上同时检测多个等位基因,且准确度高,可达到单基因的水平.Kover等[26]培育出了第1套拟南芥MAGIC系,构建了19个拟南芥亲本互交的含1026个株系的MAGIC 群体.利用该群体结合表型数据采用多种模型进行QTL作图,通过模拟发现可以检测到大部分表型变异超过10%的数量性状,证实可以利用MAGIC群体进行遗传定位分析. ...
A multiparent advanced generation inter-cross to fine-map quantitative traits in Arabidopsis thaliana
3
2009
... 目前,MAGIC群体主要基于以下2点进行遗传定位,一是利用基因型和表型数据进行关联分析,二是基于标记构建单倍型的连锁分析[25].关联分析在MAGIC群体中一般进行重要性状的遗传定位.与连锁定位相比,关联分析广度大,主要基于群体中普遍存在连锁不平衡现象,因此可以在同一座位上同时检测多个等位基因,且准确度高,可达到单基因的水平.Kover等[26]培育出了第1套拟南芥MAGIC系,构建了19个拟南芥亲本互交的含1026个株系的MAGIC 群体.利用该群体结合表型数据采用多种模型进行QTL作图,通过模拟发现可以检测到大部分表型变异超过10%的数量性状,证实可以利用MAGIC群体进行遗传定位分析. ...
... Application of MAGIC group in plant genetics research
Table 1 作物
Crop | 类型
Type | 亲本选择
Parental selection | 杂交方式
Hybridization | 群体规模
Group size | 基因型鉴定
Genotyping | 研究性状
Research trait | 分析方法
Analytical method | 参考文献
Reference |
拟南芥
Arabidopsis | –
| 19亲本
| 随机杂交
| 1026 F4
| 1260 SNPs
| 抽薹期
| QTL
| [26]
|
| 水稻Rice | 籼稻 | 4亲本DC1 | 双列杂交 | 495 F6 | 6K SNP芯片 | 籽粒产量性状、穗相关性状、分蘖相关性状、抽穗期、苗期和株高
| GWAS | [23,28-31] |
| | 4亲本DC2 | 双列杂交 | 525 F6 | | | |
| | 8亲本
| Funnel杂交
| 668 F6
| 55K SNP芯片,
GBS | | |
| 籼稻 | 8亲本 | Funnel杂交 | 1328 S7 | GBS: 17387 SNP | 产量性状、稻米品质、开花期、株高、粒型、抗旱性、耐涝性、耐盐性、抗病性(稻瘟病、白叶枯病、褐斑病)
| QTL,GWAS | [32⇓-34] |
| | MAGIC PLUS | ― | 144 S8 | | | |
| | MAGIC PLUS
DH系 | ―
| 76 DH
| | | |
| 粳稻 | 8亲本 | Funnel杂交 | 500 S5 | | | |
| 籼―粳 | GLOBAL MAGIC | ― | 1402 S7 | | | |
小麦Wheat
| 春小麦
| 4亲本
| Funnel杂交
| 1579 F6
| 826 DArTs、283SNPs、53SSRs,9K SNP芯片,90K SNP芯片 | 株高、籽粒重、
胚芽鞘长度
| LMC,QTL
| [13,35-36]
|
| 冬小麦
| 8亲本
| Funnel杂交
| 1091 F7
| 90K SNP芯片
| 产量、开花期、
病害和株高 | LMC,QTL
| [37-38]
|
| 全麦粉
| 4亲本
| Funnel杂交
| 332 F6
| SSR
| 产量性状、
抗白粉病 | QTL,LMC
| [39]
|
| 欧洲
小麦 | 60亲本
| 随机杂交
| 1000 S4
| 9K SNP芯片
| 抽穗期
| LDA,GWAS
| [40]
|
| | | | | 14个KASPar SNP | | | |
大麦
Hordeum | – | 8亲本 | Funnel杂交 | 5000 DH | 9K SNP芯片 | 开花期 | QTL | [41] |
| 8亲本 | Funnel杂交 | 642 F5 | 9K SNP芯片 | 黄斑病 | LMC,QTL | [42] |
| 棉花Cotton | 陆地棉 | 11亲本 | 随机杂交 | 547 F5 | GBS | 纤维品质 | GWAS,QTL | [43] |
| | 8亲本
| 随机杂交
| 960 MLs
| SLAF-seq
| 开花期、株型、
果枝数 | GWAS,QTL
| [44]
|
| | 11亲本 | 随机杂交 | 550 RIL | 47K SNP芯片 | 株高、茎秆质量 | GWAS,QTL | [45] |
高粱
Sorghum | –
| 29亲本
| 随机杂交
| 1000 F6
| GBS
| 株高
| GWAS,QTL
| [46]
|
作物
Crop | 类型
Type | 亲本选择
Parental selection | 杂交方式
Hybridization | 群体规模
Group size | 基因型鉴定
Genotyping | 研究性状
Research trait | 分析方法
Analytical method | 参考文献
Reference |
烟草
Tobacco | –
| 8亲本
| 双列杂交
| 600 F4
| 430K 芯片
| 株型、烟叶品质
| GWAS
| [47]
|
玉米
Maize | –
| 8亲本
| Funnel杂交
| 1636 F6
| GBS
| 开花期、株高、
穗位高 | LMC,AM,QTL | [48]
|
| | 4亲本
| Funnel杂交
| 1149 S3
| 118K SNP 芯片
| 株高、穗高、散
粉期、吐丝期 | QTL,GWAS
| [49]
|
| | 8亲本
| Funnel杂交
| 401 RIL
| 56K SNP 芯片
| 苗重、苗长和
黄萎病 | QTL,NAM
| [50]
|
| | 8亲本
| Funnel杂交
| 700F6
| 110K SNP
芯片 | 镰孢菌穗腐病
| LMC,AM,QTL | [51]
|
| | 8亲本
| Funnel杂交
| 406 RIL
| 100K SNP
芯片,GBS | 发芽率、叶绿素
和出苗期 | GWAS,QTL
| [52]
|
| | 8亲本
| Funnel杂交
| 368 MLs
| 1000K SNP芯片,GBS | 籽粒含油量
| GWAS,QTL
| [53]
|
| | 16亲本
| Funnel杂交
| 513 RIL
| 500K SNP芯片
| 株高、叶形态、
开花期、穗大小 | GWAS
| [54]
|
| | 4亲本
| Funnel杂交
| 1044 F6
| 50K SNP芯片
| 产量、吐丝期、
株高 | QTL,AM
| [55]
|
油菜
Brassica napus | 甘蓝型
| 8亲本
| 随机杂交
| 680 F6
| –
| 始花期、抗病性
| SSD
| [56]
|
Fn表示构建F1的杂交过程和F1自交过程的总代数,Sn表示F1植株自交的代数,DH(doubled haploid)表示双单倍体系,GBS(genotyping by sequencing)表示基于测序的基因分型,QTL(quantitative trait locus)表示QTL定位,LDA(linkage disequilibrium analysis)表示连锁不平衡分析,LMC(linkage map construction)表示连锁图谱构建,AM(association mapping)表示关联作图,GWAS(genome wide association study)表示全基因组关联分析,SNP(single nucleotide polymorphisms)表示单核苷酸多态性,Funnel杂交表示“漏斗”型杂交,SSR(simple sequence repeat)表示简单重复序列,SSD(single seed descent)表示单粒传法,RIL(recombinant inbred lines)表示重组自交系,ML(lines)表示自交系,NAM(nested association map)表示巢式关联图谱 ...
... 拟南芥的MAGIC群体是植物中构建较快的群体.Kover等[26]构建了第1套拟南芥MAGIC系,通过对所有的株系基因分型共得到1260个单核苷酸多态性(single nucleotide polymorphisms,SNP).对抽薹期进行QTL定位分析分别在1、4和5号染色体上检测到4个控制抽薹期的QTL,其中位于4号染色体上的QTL占表型变异最大,表明该QTL大概率是由与花期相关的FRIGIDA基因(位于0.26Mb)引起的;位于5号染色体上QTL(3.50 Mb)可能是另一个已知的影响开花时间自然变异的基因FLOWERING LOCUSC引起的.研究也发现了与种子萌芽和抽薹相关且未见报道的新QTL. ...
The genetic mechanism of heterosis utilization in maize improvement
1
2021
... 植物开花时间、株高、籽粒产量和环境适应性等重要的农艺性状,以及生物和非生物胁迫抗性等数量性状的表现均取决于多个基因之间相互作用[27].采用MAGIC群体进行QTL定位,能同时使用连锁作图和关联作图,使QTL定位达到更好的效果.尽管MAGIC群体的构建比较复杂,还需要很多的资源,但不同作物构建的群体数量在不断增加.目前,已经有许多MAGIC群体在不同作物中有报道,包括拟南芥、玉米、小麦、水稻、棉花、大麦和烟草等(表1),主要被用于复杂农艺性状和耐逆性的遗传研究. ...
Association mapping of ferrous,zinc,and aluminum tolerance at the seeding stage in Indica rice using MAGIC populations
2
2017
... Application of MAGIC group in plant genetics research
Table 1 作物
Crop | 类型
Type | 亲本选择
Parental selection | 杂交方式
Hybridization | 群体规模
Group size | 基因型鉴定
Genotyping | 研究性状
Research trait | 分析方法
Analytical method | 参考文献
Reference |
拟南芥
Arabidopsis | –
| 19亲本
| 随机杂交
| 1026 F4
| 1260 SNPs
| 抽薹期
| QTL
| [26]
|
| 水稻Rice | 籼稻 | 4亲本DC1 | 双列杂交 | 495 F6 | 6K SNP芯片 | 籽粒产量性状、穗相关性状、分蘖相关性状、抽穗期、苗期和株高
| GWAS | [23,28-31] |
| | 4亲本DC2 | 双列杂交 | 525 F6 | | | |
| | 8亲本
| Funnel杂交
| 668 F6
| 55K SNP芯片,
GBS | | |
| 籼稻 | 8亲本 | Funnel杂交 | 1328 S7 | GBS: 17387 SNP | 产量性状、稻米品质、开花期、株高、粒型、抗旱性、耐涝性、耐盐性、抗病性(稻瘟病、白叶枯病、褐斑病)
| QTL,GWAS | [32⇓-34] |
| | MAGIC PLUS | ― | 144 S8 | | | |
| | MAGIC PLUS
DH系 | ―
| 76 DH
| | | |
| 粳稻 | 8亲本 | Funnel杂交 | 500 S5 | | | |
| 籼―粳 | GLOBAL MAGIC | ― | 1402 S7 | | | |
小麦Wheat
| 春小麦
| 4亲本
| Funnel杂交
| 1579 F6
| 826 DArTs、283SNPs、53SSRs,9K SNP芯片,90K SNP芯片 | 株高、籽粒重、
胚芽鞘长度
| LMC,QTL
| [13,35-36]
|
| 冬小麦
| 8亲本
| Funnel杂交
| 1091 F7
| 90K SNP芯片
| 产量、开花期、
病害和株高 | LMC,QTL
| [37-38]
|
| 全麦粉
| 4亲本
| Funnel杂交
| 332 F6
| SSR
| 产量性状、
抗白粉病 | QTL,LMC
| [39]
|
| 欧洲
小麦 | 60亲本
| 随机杂交
| 1000 S4
| 9K SNP芯片
| 抽穗期
| LDA,GWAS
| [40]
|
| | | | | 14个KASPar SNP | | | |
大麦
Hordeum | – | 8亲本 | Funnel杂交 | 5000 DH | 9K SNP芯片 | 开花期 | QTL | [41] |
| 8亲本 | Funnel杂交 | 642 F5 | 9K SNP芯片 | 黄斑病 | LMC,QTL | [42] |
| 棉花Cotton | 陆地棉 | 11亲本 | 随机杂交 | 547 F5 | GBS | 纤维品质 | GWAS,QTL | [43] |
| | 8亲本
| 随机杂交
| 960 MLs
| SLAF-seq
| 开花期、株型、
果枝数 | GWAS,QTL
| [44]
|
| | 11亲本 | 随机杂交 | 550 RIL | 47K SNP芯片 | 株高、茎秆质量 | GWAS,QTL | [45] |
高粱
Sorghum | –
| 29亲本
| 随机杂交
| 1000 F6
| GBS
| 株高
| GWAS,QTL
| [46]
|
作物
Crop | 类型
Type | 亲本选择
Parental selection | 杂交方式
Hybridization | 群体规模
Group size | 基因型鉴定
Genotyping | 研究性状
Research trait | 分析方法
Analytical method | 参考文献
Reference |
烟草
Tobacco | –
| 8亲本
| 双列杂交
| 600 F4
| 430K 芯片
| 株型、烟叶品质
| GWAS
| [47]
|
玉米
Maize | –
| 8亲本
| Funnel杂交
| 1636 F6
| GBS
| 开花期、株高、
穗位高 | LMC,AM,QTL | [48]
|
| | 4亲本
| Funnel杂交
| 1149 S3
| 118K SNP 芯片
| 株高、穗高、散
粉期、吐丝期 | QTL,GWAS
| [49]
|
| | 8亲本
| Funnel杂交
| 401 RIL
| 56K SNP 芯片
| 苗重、苗长和
黄萎病 | QTL,NAM
| [50]
|
| | 8亲本
| Funnel杂交
| 700F6
| 110K SNP
芯片 | 镰孢菌穗腐病
| LMC,AM,QTL | [51]
|
| | 8亲本
| Funnel杂交
| 406 RIL
| 100K SNP
芯片,GBS | 发芽率、叶绿素
和出苗期 | GWAS,QTL
| [52]
|
| | 8亲本
| Funnel杂交
| 368 MLs
| 1000K SNP芯片,GBS | 籽粒含油量
| GWAS,QTL
| [53]
|
| | 16亲本
| Funnel杂交
| 513 RIL
| 500K SNP芯片
| 株高、叶形态、
开花期、穗大小 | GWAS
| [54]
|
| | 4亲本
| Funnel杂交
| 1044 F6
| 50K SNP芯片
| 产量、吐丝期、
株高 | QTL,AM
| [55]
|
油菜
Brassica napus | 甘蓝型
| 8亲本
| 随机杂交
| 680 F6
| –
| 始花期、抗病性
| SSD
| [56]
|
Fn表示构建F1的杂交过程和F1自交过程的总代数,Sn表示F1植株自交的代数,DH(doubled haploid)表示双单倍体系,GBS(genotyping by sequencing)表示基于测序的基因分型,QTL(quantitative trait locus)表示QTL定位,LDA(linkage disequilibrium analysis)表示连锁不平衡分析,LMC(linkage map construction)表示连锁图谱构建,AM(association mapping)表示关联作图,GWAS(genome wide association study)表示全基因组关联分析,SNP(single nucleotide polymorphisms)表示单核苷酸多态性,Funnel杂交表示“漏斗”型杂交,SSR(simple sequence repeat)表示简单重复序列,SSD(single seed descent)表示单粒传法,RIL(recombinant inbred lines)表示重组自交系,ML(lines)表示自交系,NAM(nested association map)表示巢式关联图谱 ...
... Meng等[28]构建了5个水稻MAGIC群体,并利用这5个水稻MAGIC群体进行了水稻苗期对铁、锌和铝毒害响应的关联分析,分别检测到21个与铁、铝胁迫相关的QTL和30个与锌胁迫相关的QTL.此外,Meng等[23,29]同时利用这些群体对水稻株高、产量、产量构成因素和抽穗期等14个农艺性状进行全基因组关联分析,得到除第7、9和11号染色体以外的9条染色体上共检测到26个显著的QTL定位.Ponce等[30]分别利用2个4亲本和8亲本的籼稻MAGIC群体,使用55K SNP和GBS(genotyping by sequencing)测序分别进行了基因分型,鉴定与粒长、粒宽、粒长宽比、粒厚和千粒重相关的QTL和SNP.结果显示,5个与粒级相关的性状共鉴定出18个QTL,解释了6.43%~63.35%的总表型方差.其中12个QTL与克隆基因GS3、GW5/qSW5、GW7/GL7/SLG7和GW8/OsSPL16定位一致,前2个基因对粒长和粒宽的影响最大.此外,MAGIC群体也被用于对水稻单倍体等位基因挖掘、稻米品质的QTL定位分析和生物强化等研究中. ...
Characterization of three Indica rice multiparent advanced generation inter-cross (magic) populations for quantitative trait loci (QTL) identification
1
2016
... Meng等[28]构建了5个水稻MAGIC群体,并利用这5个水稻MAGIC群体进行了水稻苗期对铁、锌和铝毒害响应的关联分析,分别检测到21个与铁、铝胁迫相关的QTL和30个与锌胁迫相关的QTL.此外,Meng等[23,29]同时利用这些群体对水稻株高、产量、产量构成因素和抽穗期等14个农艺性状进行全基因组关联分析,得到除第7、9和11号染色体以外的9条染色体上共检测到26个显著的QTL定位.Ponce等[30]分别利用2个4亲本和8亲本的籼稻MAGIC群体,使用55K SNP和GBS(genotyping by sequencing)测序分别进行了基因分型,鉴定与粒长、粒宽、粒长宽比、粒厚和千粒重相关的QTL和SNP.结果显示,5个与粒级相关的性状共鉴定出18个QTL,解释了6.43%~63.35%的总表型方差.其中12个QTL与克隆基因GS3、GW5/qSW5、GW7/GL7/SLG7和GW8/OsSPL16定位一致,前2个基因对粒长和粒宽的影响最大.此外,MAGIC群体也被用于对水稻单倍体等位基因挖掘、稻米品质的QTL定位分析和生物强化等研究中. ...
Genome-wide association study of grain size traits in Indica rice multiparent advanced generation intercross (MAGIC) population
1
2020
... Meng等[28]构建了5个水稻MAGIC群体,并利用这5个水稻MAGIC群体进行了水稻苗期对铁、锌和铝毒害响应的关联分析,分别检测到21个与铁、铝胁迫相关的QTL和30个与锌胁迫相关的QTL.此外,Meng等[23,29]同时利用这些群体对水稻株高、产量、产量构成因素和抽穗期等14个农艺性状进行全基因组关联分析,得到除第7、9和11号染色体以外的9条染色体上共检测到26个显著的QTL定位.Ponce等[30]分别利用2个4亲本和8亲本的籼稻MAGIC群体,使用55K SNP和GBS(genotyping by sequencing)测序分别进行了基因分型,鉴定与粒长、粒宽、粒长宽比、粒厚和千粒重相关的QTL和SNP.结果显示,5个与粒级相关的性状共鉴定出18个QTL,解释了6.43%~63.35%的总表型方差.其中12个QTL与克隆基因GS3、GW5/qSW5、GW7/GL7/SLG7和GW8/OsSPL16定位一致,前2个基因对粒长和粒宽的影响最大.此外,MAGIC群体也被用于对水稻单倍体等位基因挖掘、稻米品质的QTL定位分析和生物强化等研究中. ...
Effciently tracking selection in a multiparental population:the case of earliness in wheat
1
2015
... Application of MAGIC group in plant genetics research
Table 1 作物
Crop | 类型
Type | 亲本选择
Parental selection | 杂交方式
Hybridization | 群体规模
Group size | 基因型鉴定
Genotyping | 研究性状
Research trait | 分析方法
Analytical method | 参考文献
Reference |
拟南芥
Arabidopsis | –
| 19亲本
| 随机杂交
| 1026 F4
| 1260 SNPs
| 抽薹期
| QTL
| [26]
|
| 水稻Rice | 籼稻 | 4亲本DC1 | 双列杂交 | 495 F6 | 6K SNP芯片 | 籽粒产量性状、穗相关性状、分蘖相关性状、抽穗期、苗期和株高
| GWAS | [23,28-31] |
| | 4亲本DC2 | 双列杂交 | 525 F6 | | | |
| | 8亲本
| Funnel杂交
| 668 F6
| 55K SNP芯片,
GBS | | |
| 籼稻 | 8亲本 | Funnel杂交 | 1328 S7 | GBS: 17387 SNP | 产量性状、稻米品质、开花期、株高、粒型、抗旱性、耐涝性、耐盐性、抗病性(稻瘟病、白叶枯病、褐斑病)
| QTL,GWAS | [32⇓-34] |
| | MAGIC PLUS | ― | 144 S8 | | | |
| | MAGIC PLUS
DH系 | ―
| 76 DH
| | | |
| 粳稻 | 8亲本 | Funnel杂交 | 500 S5 | | | |
| 籼―粳 | GLOBAL MAGIC | ― | 1402 S7 | | | |
小麦Wheat
| 春小麦
| 4亲本
| Funnel杂交
| 1579 F6
| 826 DArTs、283SNPs、53SSRs,9K SNP芯片,90K SNP芯片 | 株高、籽粒重、
胚芽鞘长度
| LMC,QTL
| [13,35-36]
|
| 冬小麦
| 8亲本
| Funnel杂交
| 1091 F7
| 90K SNP芯片
| 产量、开花期、
病害和株高 | LMC,QTL
| [37-38]
|
| 全麦粉
| 4亲本
| Funnel杂交
| 332 F6
| SSR
| 产量性状、
抗白粉病 | QTL,LMC
| [39]
|
| 欧洲
小麦 | 60亲本
| 随机杂交
| 1000 S4
| 9K SNP芯片
| 抽穗期
| LDA,GWAS
| [40]
|
| | | | | 14个KASPar SNP | | | |
大麦
Hordeum | – | 8亲本 | Funnel杂交 | 5000 DH | 9K SNP芯片 | 开花期 | QTL | [41] |
| 8亲本 | Funnel杂交 | 642 F5 | 9K SNP芯片 | 黄斑病 | LMC,QTL | [42] |
| 棉花Cotton | 陆地棉 | 11亲本 | 随机杂交 | 547 F5 | GBS | 纤维品质 | GWAS,QTL | [43] |
| | 8亲本
| 随机杂交
| 960 MLs
| SLAF-seq
| 开花期、株型、
果枝数 | GWAS,QTL
| [44]
|
| | 11亲本 | 随机杂交 | 550 RIL | 47K SNP芯片 | 株高、茎秆质量 | GWAS,QTL | [45] |
高粱
Sorghum | –
| 29亲本
| 随机杂交
| 1000 F6
| GBS
| 株高
| GWAS,QTL
| [46]
|
作物
Crop | 类型
Type | 亲本选择
Parental selection | 杂交方式
Hybridization | 群体规模
Group size | 基因型鉴定
Genotyping | 研究性状
Research trait | 分析方法
Analytical method | 参考文献
Reference |
烟草
Tobacco | –
| 8亲本
| 双列杂交
| 600 F4
| 430K 芯片
| 株型、烟叶品质
| GWAS
| [47]
|
玉米
Maize | –
| 8亲本
| Funnel杂交
| 1636 F6
| GBS
| 开花期、株高、
穗位高 | LMC,AM,QTL | [48]
|
| | 4亲本
| Funnel杂交
| 1149 S3
| 118K SNP 芯片
| 株高、穗高、散
粉期、吐丝期 | QTL,GWAS
| [49]
|
| | 8亲本
| Funnel杂交
| 401 RIL
| 56K SNP 芯片
| 苗重、苗长和
黄萎病 | QTL,NAM
| [50]
|
| | 8亲本
| Funnel杂交
| 700F6
| 110K SNP
芯片 | 镰孢菌穗腐病
| LMC,AM,QTL | [51]
|
| | 8亲本
| Funnel杂交
| 406 RIL
| 100K SNP
芯片,GBS | 发芽率、叶绿素
和出苗期 | GWAS,QTL
| [52]
|
| | 8亲本
| Funnel杂交
| 368 MLs
| 1000K SNP芯片,GBS | 籽粒含油量
| GWAS,QTL
| [53]
|
| | 16亲本
| Funnel杂交
| 513 RIL
| 500K SNP芯片
| 株高、叶形态、
开花期、穗大小 | GWAS
| [54]
|
| | 4亲本
| Funnel杂交
| 1044 F6
| 50K SNP芯片
| 产量、吐丝期、
株高 | QTL,AM
| [55]
|
油菜
Brassica napus | 甘蓝型
| 8亲本
| 随机杂交
| 680 F6
| –
| 始花期、抗病性
| SSD
| [56]
|
Fn表示构建F1的杂交过程和F1自交过程的总代数,Sn表示F1植株自交的代数,DH(doubled haploid)表示双单倍体系,GBS(genotyping by sequencing)表示基于测序的基因分型,QTL(quantitative trait locus)表示QTL定位,LDA(linkage disequilibrium analysis)表示连锁不平衡分析,LMC(linkage map construction)表示连锁图谱构建,AM(association mapping)表示关联作图,GWAS(genome wide association study)表示全基因组关联分析,SNP(single nucleotide polymorphisms)表示单核苷酸多态性,Funnel杂交表示“漏斗”型杂交,SSR(simple sequence repeat)表示简单重复序列,SSD(single seed descent)表示单粒传法,RIL(recombinant inbred lines)表示重组自交系,ML(lines)表示自交系,NAM(nested association map)表示巢式关联图谱 ...
Multi-parent advanced generation inter-cross (MAGIC) populations in rice:progress and potential for genetics research and breeding
2
2013
... Application of MAGIC group in plant genetics research
Table 1 作物
Crop | 类型
Type | 亲本选择
Parental selection | 杂交方式
Hybridization | 群体规模
Group size | 基因型鉴定
Genotyping | 研究性状
Research trait | 分析方法
Analytical method | 参考文献
Reference |
拟南芥
Arabidopsis | –
| 19亲本
| 随机杂交
| 1026 F4
| 1260 SNPs
| 抽薹期
| QTL
| [26]
|
| 水稻Rice | 籼稻 | 4亲本DC1 | 双列杂交 | 495 F6 | 6K SNP芯片 | 籽粒产量性状、穗相关性状、分蘖相关性状、抽穗期、苗期和株高
| GWAS | [23,28-31] |
| | 4亲本DC2 | 双列杂交 | 525 F6 | | | |
| | 8亲本
| Funnel杂交
| 668 F6
| 55K SNP芯片,
GBS | | |
| 籼稻 | 8亲本 | Funnel杂交 | 1328 S7 | GBS: 17387 SNP | 产量性状、稻米品质、开花期、株高、粒型、抗旱性、耐涝性、耐盐性、抗病性(稻瘟病、白叶枯病、褐斑病)
| QTL,GWAS | [32⇓-34] |
| | MAGIC PLUS | ― | 144 S8 | | | |
| | MAGIC PLUS
DH系 | ―
| 76 DH
| | | |
| 粳稻 | 8亲本 | Funnel杂交 | 500 S5 | | | |
| 籼―粳 | GLOBAL MAGIC | ― | 1402 S7 | | | |
小麦Wheat
| 春小麦
| 4亲本
| Funnel杂交
| 1579 F6
| 826 DArTs、283SNPs、53SSRs,9K SNP芯片,90K SNP芯片 | 株高、籽粒重、
胚芽鞘长度
| LMC,QTL
| [13,35-36]
|
| 冬小麦
| 8亲本
| Funnel杂交
| 1091 F7
| 90K SNP芯片
| 产量、开花期、
病害和株高 | LMC,QTL
| [37-38]
|
| 全麦粉
| 4亲本
| Funnel杂交
| 332 F6
| SSR
| 产量性状、
抗白粉病 | QTL,LMC
| [39]
|
| 欧洲
小麦 | 60亲本
| 随机杂交
| 1000 S4
| 9K SNP芯片
| 抽穗期
| LDA,GWAS
| [40]
|
| | | | | 14个KASPar SNP | | | |
大麦
Hordeum | – | 8亲本 | Funnel杂交 | 5000 DH | 9K SNP芯片 | 开花期 | QTL | [41] |
| 8亲本 | Funnel杂交 | 642 F5 | 9K SNP芯片 | 黄斑病 | LMC,QTL | [42] |
| 棉花Cotton | 陆地棉 | 11亲本 | 随机杂交 | 547 F5 | GBS | 纤维品质 | GWAS,QTL | [43] |
| | 8亲本
| 随机杂交
| 960 MLs
| SLAF-seq
| 开花期、株型、
果枝数 | GWAS,QTL
| [44]
|
| | 11亲本 | 随机杂交 | 550 RIL | 47K SNP芯片 | 株高、茎秆质量 | GWAS,QTL | [45] |
高粱
Sorghum | –
| 29亲本
| 随机杂交
| 1000 F6
| GBS
| 株高
| GWAS,QTL
| [46]
|
作物
Crop | 类型
Type | 亲本选择
Parental selection | 杂交方式
Hybridization | 群体规模
Group size | 基因型鉴定
Genotyping | 研究性状
Research trait | 分析方法
Analytical method | 参考文献
Reference |
烟草
Tobacco | –
| 8亲本
| 双列杂交
| 600 F4
| 430K 芯片
| 株型、烟叶品质
| GWAS
| [47]
|
玉米
Maize | –
| 8亲本
| Funnel杂交
| 1636 F6
| GBS
| 开花期、株高、
穗位高 | LMC,AM,QTL | [48]
|
| | 4亲本
| Funnel杂交
| 1149 S3
| 118K SNP 芯片
| 株高、穗高、散
粉期、吐丝期 | QTL,GWAS
| [49]
|
| | 8亲本
| Funnel杂交
| 401 RIL
| 56K SNP 芯片
| 苗重、苗长和
黄萎病 | QTL,NAM
| [50]
|
| | 8亲本
| Funnel杂交
| 700F6
| 110K SNP
芯片 | 镰孢菌穗腐病
| LMC,AM,QTL | [51]
|
| | 8亲本
| Funnel杂交
| 406 RIL
| 100K SNP
芯片,GBS | 发芽率、叶绿素
和出苗期 | GWAS,QTL
| [52]
|
| | 8亲本
| Funnel杂交
| 368 MLs
| 1000K SNP芯片,GBS | 籽粒含油量
| GWAS,QTL
| [53]
|
| | 16亲本
| Funnel杂交
| 513 RIL
| 500K SNP芯片
| 株高、叶形态、
开花期、穗大小 | GWAS
| [54]
|
| | 4亲本
| Funnel杂交
| 1044 F6
| 50K SNP芯片
| 产量、吐丝期、
株高 | QTL,AM
| [55]
|
油菜
Brassica napus | 甘蓝型
| 8亲本
| 随机杂交
| 680 F6
| –
| 始花期、抗病性
| SSD
| [56]
|
Fn表示构建F1的杂交过程和F1自交过程的总代数,Sn表示F1植株自交的代数,DH(doubled haploid)表示双单倍体系,GBS(genotyping by sequencing)表示基于测序的基因分型,QTL(quantitative trait locus)表示QTL定位,LDA(linkage disequilibrium analysis)表示连锁不平衡分析,LMC(linkage map construction)表示连锁图谱构建,AM(association mapping)表示关联作图,GWAS(genome wide association study)表示全基因组关联分析,SNP(single nucleotide polymorphisms)表示单核苷酸多态性,Funnel杂交表示“漏斗”型杂交,SSR(simple sequence repeat)表示简单重复序列,SSD(single seed descent)表示单粒传法,RIL(recombinant inbred lines)表示重组自交系,ML(lines)表示自交系,NAM(nested association map)表示巢式关联图谱 ...
... 水稻MAGIC群体被应用于分析多种表型,其中研究最多的是复杂性状,如产量、耐旱和抗病性等.Bandillo等[32]构建了4个多亲本的群体,包括8亲本的粳稻MAGIC群体,8亲本的籼稻MAGIC群体,8亲本的籼稻MAGIC plus群体和16亲本(8个粳稻亲本和8个籼稻亲本)的Global MAGIC群体,并利用这些群体对水稻的抗稻瘟病、抗白叶枯病、耐盐性和耐涝性的稻米品质进行了全基因组关联分析,成功定位到显著性相关位点,包括与耐涝相关的基因Sub1和与抗白叶枯病相关的基因Xa4和Xa5.陈天晓等[33]和Yu等[62]利用8个具有丰富多样性的优良种质为亲本构建了2个8亲本和2个4亲本MAGIC群体.在这3个MAGIC群体分别接种GD-V(强致病菌)和C2(弱致病菌)致病力不同的水稻白叶枯病菌,对水稻抗白叶枯病进行QTL定位研究和群体结构分析,在3个群体917个稳定株系中共得到3128个高质量的SNP位点. ...
利用水稻MAGIC群体关联定位白叶枯病抗性QTL和创制抗病新种质
2
2016
... Application of MAGIC group in plant genetics research
Table 1 作物
Crop | 类型
Type | 亲本选择
Parental selection | 杂交方式
Hybridization | 群体规模
Group size | 基因型鉴定
Genotyping | 研究性状
Research trait | 分析方法
Analytical method | 参考文献
Reference |
拟南芥
Arabidopsis | –
| 19亲本
| 随机杂交
| 1026 F4
| 1260 SNPs
| 抽薹期
| QTL
| [26]
|
| 水稻Rice | 籼稻 | 4亲本DC1 | 双列杂交 | 495 F6 | 6K SNP芯片 | 籽粒产量性状、穗相关性状、分蘖相关性状、抽穗期、苗期和株高
| GWAS | [23,28-31] |
| | 4亲本DC2 | 双列杂交 | 525 F6 | | | |
| | 8亲本
| Funnel杂交
| 668 F6
| 55K SNP芯片,
GBS | | |
| 籼稻 | 8亲本 | Funnel杂交 | 1328 S7 | GBS: 17387 SNP | 产量性状、稻米品质、开花期、株高、粒型、抗旱性、耐涝性、耐盐性、抗病性(稻瘟病、白叶枯病、褐斑病)
| QTL,GWAS | [32⇓-34] |
| | MAGIC PLUS | ― | 144 S8 | | | |
| | MAGIC PLUS
DH系 | ―
| 76 DH
| | | |
| 粳稻 | 8亲本 | Funnel杂交 | 500 S5 | | | |
| 籼―粳 | GLOBAL MAGIC | ― | 1402 S7 | | | |
小麦Wheat
| 春小麦
| 4亲本
| Funnel杂交
| 1579 F6
| 826 DArTs、283SNPs、53SSRs,9K SNP芯片,90K SNP芯片 | 株高、籽粒重、
胚芽鞘长度
| LMC,QTL
| [13,35-36]
|
| 冬小麦
| 8亲本
| Funnel杂交
| 1091 F7
| 90K SNP芯片
| 产量、开花期、
病害和株高 | LMC,QTL
| [37-38]
|
| 全麦粉
| 4亲本
| Funnel杂交
| 332 F6
| SSR
| 产量性状、
抗白粉病 | QTL,LMC
| [39]
|
| 欧洲
小麦 | 60亲本
| 随机杂交
| 1000 S4
| 9K SNP芯片
| 抽穗期
| LDA,GWAS
| [40]
|
| | | | | 14个KASPar SNP | | | |
大麦
Hordeum | – | 8亲本 | Funnel杂交 | 5000 DH | 9K SNP芯片 | 开花期 | QTL | [41] |
| 8亲本 | Funnel杂交 | 642 F5 | 9K SNP芯片 | 黄斑病 | LMC,QTL | [42] |
| 棉花Cotton | 陆地棉 | 11亲本 | 随机杂交 | 547 F5 | GBS | 纤维品质 | GWAS,QTL | [43] |
| | 8亲本
| 随机杂交
| 960 MLs
| SLAF-seq
| 开花期、株型、
果枝数 | GWAS,QTL
| [44]
|
| | 11亲本 | 随机杂交 | 550 RIL | 47K SNP芯片 | 株高、茎秆质量 | GWAS,QTL | [45] |
高粱
Sorghum | –
| 29亲本
| 随机杂交
| 1000 F6
| GBS
| 株高
| GWAS,QTL
| [46]
|
作物
Crop | 类型
Type | 亲本选择
Parental selection | 杂交方式
Hybridization | 群体规模
Group size | 基因型鉴定
Genotyping | 研究性状
Research trait | 分析方法
Analytical method | 参考文献
Reference |
烟草
Tobacco | –
| 8亲本
| 双列杂交
| 600 F4
| 430K 芯片
| 株型、烟叶品质
| GWAS
| [47]
|
玉米
Maize | –
| 8亲本
| Funnel杂交
| 1636 F6
| GBS
| 开花期、株高、
穗位高 | LMC,AM,QTL | [48]
|
| | 4亲本
| Funnel杂交
| 1149 S3
| 118K SNP 芯片
| 株高、穗高、散
粉期、吐丝期 | QTL,GWAS
| [49]
|
| | 8亲本
| Funnel杂交
| 401 RIL
| 56K SNP 芯片
| 苗重、苗长和
黄萎病 | QTL,NAM
| [50]
|
| | 8亲本
| Funnel杂交
| 700F6
| 110K SNP
芯片 | 镰孢菌穗腐病
| LMC,AM,QTL | [51]
|
| | 8亲本
| Funnel杂交
| 406 RIL
| 100K SNP
芯片,GBS | 发芽率、叶绿素
和出苗期 | GWAS,QTL
| [52]
|
| | 8亲本
| Funnel杂交
| 368 MLs
| 1000K SNP芯片,GBS | 籽粒含油量
| GWAS,QTL
| [53]
|
| | 16亲本
| Funnel杂交
| 513 RIL
| 500K SNP芯片
| 株高、叶形态、
开花期、穗大小 | GWAS
| [54]
|
| | 4亲本
| Funnel杂交
| 1044 F6
| 50K SNP芯片
| 产量、吐丝期、
株高 | QTL,AM
| [55]
|
油菜
Brassica napus | 甘蓝型
| 8亲本
| 随机杂交
| 680 F6
| –
| 始花期、抗病性
| SSD
| [56]
|
Fn表示构建F1的杂交过程和F1自交过程的总代数,Sn表示F1植株自交的代数,DH(doubled haploid)表示双单倍体系,GBS(genotyping by sequencing)表示基于测序的基因分型,QTL(quantitative trait locus)表示QTL定位,LDA(linkage disequilibrium analysis)表示连锁不平衡分析,LMC(linkage map construction)表示连锁图谱构建,AM(association mapping)表示关联作图,GWAS(genome wide association study)表示全基因组关联分析,SNP(single nucleotide polymorphisms)表示单核苷酸多态性,Funnel杂交表示“漏斗”型杂交,SSR(simple sequence repeat)表示简单重复序列,SSD(single seed descent)表示单粒传法,RIL(recombinant inbred lines)表示重组自交系,ML(lines)表示自交系,NAM(nested association map)表示巢式关联图谱 ...
... 水稻MAGIC群体被应用于分析多种表型,其中研究最多的是复杂性状,如产量、耐旱和抗病性等.Bandillo等[32]构建了4个多亲本的群体,包括8亲本的粳稻MAGIC群体,8亲本的籼稻MAGIC群体,8亲本的籼稻MAGIC plus群体和16亲本(8个粳稻亲本和8个籼稻亲本)的Global MAGIC群体,并利用这些群体对水稻的抗稻瘟病、抗白叶枯病、耐盐性和耐涝性的稻米品质进行了全基因组关联分析,成功定位到显著性相关位点,包括与耐涝相关的基因Sub1和与抗白叶枯病相关的基因Xa4和Xa5.陈天晓等[33]和Yu等[62]利用8个具有丰富多样性的优良种质为亲本构建了2个8亲本和2个4亲本MAGIC群体.在这3个MAGIC群体分别接种GD-V(强致病菌)和C2(弱致病菌)致病力不同的水稻白叶枯病菌,对水稻抗白叶枯病进行QTL定位研究和群体结构分析,在3个群体917个稳定株系中共得到3128个高质量的SNP位点. ...
Approaches in characterizing genetic structure and mapping in a rice multiparental population
1
2017
... Application of MAGIC group in plant genetics research
Table 1 作物
Crop | 类型
Type | 亲本选择
Parental selection | 杂交方式
Hybridization | 群体规模
Group size | 基因型鉴定
Genotyping | 研究性状
Research trait | 分析方法
Analytical method | 参考文献
Reference |
拟南芥
Arabidopsis | –
| 19亲本
| 随机杂交
| 1026 F4
| 1260 SNPs
| 抽薹期
| QTL
| [26]
|
| 水稻Rice | 籼稻 | 4亲本DC1 | 双列杂交 | 495 F6 | 6K SNP芯片 | 籽粒产量性状、穗相关性状、分蘖相关性状、抽穗期、苗期和株高
| GWAS | [23,28-31] |
| | 4亲本DC2 | 双列杂交 | 525 F6 | | | |
| | 8亲本
| Funnel杂交
| 668 F6
| 55K SNP芯片,
GBS | | |
| 籼稻 | 8亲本 | Funnel杂交 | 1328 S7 | GBS: 17387 SNP | 产量性状、稻米品质、开花期、株高、粒型、抗旱性、耐涝性、耐盐性、抗病性(稻瘟病、白叶枯病、褐斑病)
| QTL,GWAS | [32⇓-34] |
| | MAGIC PLUS | ― | 144 S8 | | | |
| | MAGIC PLUS
DH系 | ―
| 76 DH
| | | |
| 粳稻 | 8亲本 | Funnel杂交 | 500 S5 | | | |
| 籼―粳 | GLOBAL MAGIC | ― | 1402 S7 | | | |
小麦Wheat
| 春小麦
| 4亲本
| Funnel杂交
| 1579 F6
| 826 DArTs、283SNPs、53SSRs,9K SNP芯片,90K SNP芯片 | 株高、籽粒重、
胚芽鞘长度
| LMC,QTL
| [13,35-36]
|
| 冬小麦
| 8亲本
| Funnel杂交
| 1091 F7
| 90K SNP芯片
| 产量、开花期、
病害和株高 | LMC,QTL
| [37-38]
|
| 全麦粉
| 4亲本
| Funnel杂交
| 332 F6
| SSR
| 产量性状、
抗白粉病 | QTL,LMC
| [39]
|
| 欧洲
小麦 | 60亲本
| 随机杂交
| 1000 S4
| 9K SNP芯片
| 抽穗期
| LDA,GWAS
| [40]
|
| | | | | 14个KASPar SNP | | | |
大麦
Hordeum | – | 8亲本 | Funnel杂交 | 5000 DH | 9K SNP芯片 | 开花期 | QTL | [41] |
| 8亲本 | Funnel杂交 | 642 F5 | 9K SNP芯片 | 黄斑病 | LMC,QTL | [42] |
| 棉花Cotton | 陆地棉 | 11亲本 | 随机杂交 | 547 F5 | GBS | 纤维品质 | GWAS,QTL | [43] |
| | 8亲本
| 随机杂交
| 960 MLs
| SLAF-seq
| 开花期、株型、
果枝数 | GWAS,QTL
| [44]
|
| | 11亲本 | 随机杂交 | 550 RIL | 47K SNP芯片 | 株高、茎秆质量 | GWAS,QTL | [45] |
高粱
Sorghum | –
| 29亲本
| 随机杂交
| 1000 F6
| GBS
| 株高
| GWAS,QTL
| [46]
|
作物
Crop | 类型
Type | 亲本选择
Parental selection | 杂交方式
Hybridization | 群体规模
Group size | 基因型鉴定
Genotyping | 研究性状
Research trait | 分析方法
Analytical method | 参考文献
Reference |
烟草
Tobacco | –
| 8亲本
| 双列杂交
| 600 F4
| 430K 芯片
| 株型、烟叶品质
| GWAS
| [47]
|
玉米
Maize | –
| 8亲本
| Funnel杂交
| 1636 F6
| GBS
| 开花期、株高、
穗位高 | LMC,AM,QTL | [48]
|
| | 4亲本
| Funnel杂交
| 1149 S3
| 118K SNP 芯片
| 株高、穗高、散
粉期、吐丝期 | QTL,GWAS
| [49]
|
| | 8亲本
| Funnel杂交
| 401 RIL
| 56K SNP 芯片
| 苗重、苗长和
黄萎病 | QTL,NAM
| [50]
|
| | 8亲本
| Funnel杂交
| 700F6
| 110K SNP
芯片 | 镰孢菌穗腐病
| LMC,AM,QTL | [51]
|
| | 8亲本
| Funnel杂交
| 406 RIL
| 100K SNP
芯片,GBS | 发芽率、叶绿素
和出苗期 | GWAS,QTL
| [52]
|
| | 8亲本
| Funnel杂交
| 368 MLs
| 1000K SNP芯片,GBS | 籽粒含油量
| GWAS,QTL
| [53]
|
| | 16亲本
| Funnel杂交
| 513 RIL
| 500K SNP芯片
| 株高、叶形态、
开花期、穗大小 | GWAS
| [54]
|
| | 4亲本
| Funnel杂交
| 1044 F6
| 50K SNP芯片
| 产量、吐丝期、
株高 | QTL,AM
| [55]
|
油菜
Brassica napus | 甘蓝型
| 8亲本
| 随机杂交
| 680 F6
| –
| 始花期、抗病性
| SSD
| [56]
|
Fn表示构建F1的杂交过程和F1自交过程的总代数,Sn表示F1植株自交的代数,DH(doubled haploid)表示双单倍体系,GBS(genotyping by sequencing)表示基于测序的基因分型,QTL(quantitative trait locus)表示QTL定位,LDA(linkage disequilibrium analysis)表示连锁不平衡分析,LMC(linkage map construction)表示连锁图谱构建,AM(association mapping)表示关联作图,GWAS(genome wide association study)表示全基因组关联分析,SNP(single nucleotide polymorphisms)表示单核苷酸多态性,Funnel杂交表示“漏斗”型杂交,SSR(simple sequence repeat)表示简单重复序列,SSD(single seed descent)表示单粒传法,RIL(recombinant inbred lines)表示重组自交系,ML(lines)表示自交系,NAM(nested association map)表示巢式关联图谱 ...
A multiparent advanced generation inter-cross population for genetic analysis in wheat
2
2012
... Application of MAGIC group in plant genetics research
Table 1 作物
Crop | 类型
Type | 亲本选择
Parental selection | 杂交方式
Hybridization | 群体规模
Group size | 基因型鉴定
Genotyping | 研究性状
Research trait | 分析方法
Analytical method | 参考文献
Reference |
拟南芥
Arabidopsis | –
| 19亲本
| 随机杂交
| 1026 F4
| 1260 SNPs
| 抽薹期
| QTL
| [26]
|
| 水稻Rice | 籼稻 | 4亲本DC1 | 双列杂交 | 495 F6 | 6K SNP芯片 | 籽粒产量性状、穗相关性状、分蘖相关性状、抽穗期、苗期和株高
| GWAS | [23,28-31] |
| | 4亲本DC2 | 双列杂交 | 525 F6 | | | |
| | 8亲本
| Funnel杂交
| 668 F6
| 55K SNP芯片,
GBS | | |
| 籼稻 | 8亲本 | Funnel杂交 | 1328 S7 | GBS: 17387 SNP | 产量性状、稻米品质、开花期、株高、粒型、抗旱性、耐涝性、耐盐性、抗病性(稻瘟病、白叶枯病、褐斑病)
| QTL,GWAS | [32⇓-34] |
| | MAGIC PLUS | ― | 144 S8 | | | |
| | MAGIC PLUS
DH系 | ―
| 76 DH
| | | |
| 粳稻 | 8亲本 | Funnel杂交 | 500 S5 | | | |
| 籼―粳 | GLOBAL MAGIC | ― | 1402 S7 | | | |
小麦Wheat
| 春小麦
| 4亲本
| Funnel杂交
| 1579 F6
| 826 DArTs、283SNPs、53SSRs,9K SNP芯片,90K SNP芯片 | 株高、籽粒重、
胚芽鞘长度
| LMC,QTL
| [13,35-36]
|
| 冬小麦
| 8亲本
| Funnel杂交
| 1091 F7
| 90K SNP芯片
| 产量、开花期、
病害和株高 | LMC,QTL
| [37-38]
|
| 全麦粉
| 4亲本
| Funnel杂交
| 332 F6
| SSR
| 产量性状、
抗白粉病 | QTL,LMC
| [39]
|
| 欧洲
小麦 | 60亲本
| 随机杂交
| 1000 S4
| 9K SNP芯片
| 抽穗期
| LDA,GWAS
| [40]
|
| | | | | 14个KASPar SNP | | | |
大麦
Hordeum | – | 8亲本 | Funnel杂交 | 5000 DH | 9K SNP芯片 | 开花期 | QTL | [41] |
| 8亲本 | Funnel杂交 | 642 F5 | 9K SNP芯片 | 黄斑病 | LMC,QTL | [42] |
| 棉花Cotton | 陆地棉 | 11亲本 | 随机杂交 | 547 F5 | GBS | 纤维品质 | GWAS,QTL | [43] |
| | 8亲本
| 随机杂交
| 960 MLs
| SLAF-seq
| 开花期、株型、
果枝数 | GWAS,QTL
| [44]
|
| | 11亲本 | 随机杂交 | 550 RIL | 47K SNP芯片 | 株高、茎秆质量 | GWAS,QTL | [45] |
高粱
Sorghum | –
| 29亲本
| 随机杂交
| 1000 F6
| GBS
| 株高
| GWAS,QTL
| [46]
|
作物
Crop | 类型
Type | 亲本选择
Parental selection | 杂交方式
Hybridization | 群体规模
Group size | 基因型鉴定
Genotyping | 研究性状
Research trait | 分析方法
Analytical method | 参考文献
Reference |
烟草
Tobacco | –
| 8亲本
| 双列杂交
| 600 F4
| 430K 芯片
| 株型、烟叶品质
| GWAS
| [47]
|
玉米
Maize | –
| 8亲本
| Funnel杂交
| 1636 F6
| GBS
| 开花期、株高、
穗位高 | LMC,AM,QTL | [48]
|
| | 4亲本
| Funnel杂交
| 1149 S3
| 118K SNP 芯片
| 株高、穗高、散
粉期、吐丝期 | QTL,GWAS
| [49]
|
| | 8亲本
| Funnel杂交
| 401 RIL
| 56K SNP 芯片
| 苗重、苗长和
黄萎病 | QTL,NAM
| [50]
|
| | 8亲本
| Funnel杂交
| 700F6
| 110K SNP
芯片 | 镰孢菌穗腐病
| LMC,AM,QTL | [51]
|
| | 8亲本
| Funnel杂交
| 406 RIL
| 100K SNP
芯片,GBS | 发芽率、叶绿素
和出苗期 | GWAS,QTL
| [52]
|
| | 8亲本
| Funnel杂交
| 368 MLs
| 1000K SNP芯片,GBS | 籽粒含油量
| GWAS,QTL
| [53]
|
| | 16亲本
| Funnel杂交
| 513 RIL
| 500K SNP芯片
| 株高、叶形态、
开花期、穗大小 | GWAS
| [54]
|
| | 4亲本
| Funnel杂交
| 1044 F6
| 50K SNP芯片
| 产量、吐丝期、
株高 | QTL,AM
| [55]
|
油菜
Brassica napus | 甘蓝型
| 8亲本
| 随机杂交
| 680 F6
| –
| 始花期、抗病性
| SSD
| [56]
|
Fn表示构建F1的杂交过程和F1自交过程的总代数,Sn表示F1植株自交的代数,DH(doubled haploid)表示双单倍体系,GBS(genotyping by sequencing)表示基于测序的基因分型,QTL(quantitative trait locus)表示QTL定位,LDA(linkage disequilibrium analysis)表示连锁不平衡分析,LMC(linkage map construction)表示连锁图谱构建,AM(association mapping)表示关联作图,GWAS(genome wide association study)表示全基因组关联分析,SNP(single nucleotide polymorphisms)表示单核苷酸多态性,Funnel杂交表示“漏斗”型杂交,SSR(simple sequence repeat)表示简单重复序列,SSD(single seed descent)表示单粒传法,RIL(recombinant inbred lines)表示重组自交系,ML(lines)表示自交系,NAM(nested association map)表示巢式关联图谱 ...
... 小麦基因组结构复杂,对于MAGIC群体也进行了深入遗传研究.第1个小麦MAGIC群体是Huang等[35]使用4个澳洲春小麦作为亲本构建了包含1579个 RILs的MAGIC群体.利用1162个DArT(diversity arrays technology)、SNP和SSR(simple sequence repeat)标记鉴定了871个家系的基因型并且构建了遗传图谱,检测到9个株高和8个百粒重的QTL,其中有3个株高QTL和已知基因相邻.Mackay等[37,57]采用8个冬小麦亲本构建了1个包含1091个家系的MAGIC群体.通过使用Illumina平台的90 000 SNP小麦芯片鉴定720个家系和8个亲本的基因型,共获得62 543个SNP标记.他们利用SNP标记通过连锁不平衡分析了基因组重组.小麦MAGIC群体是数据收集中进展最快的群体,推动了小麦遗传分析方法的发展,无论是在连锁图谱构建领域,还是标记和性状的关联定位,都取得了很好的进展. ...
Use of a large multiparent wheat mapping population in genomic dissection of coleoptile and seedling growth
1
2014
... Application of MAGIC group in plant genetics research
Table 1 作物
Crop | 类型
Type | 亲本选择
Parental selection | 杂交方式
Hybridization | 群体规模
Group size | 基因型鉴定
Genotyping | 研究性状
Research trait | 分析方法
Analytical method | 参考文献
Reference |
拟南芥
Arabidopsis | –
| 19亲本
| 随机杂交
| 1026 F4
| 1260 SNPs
| 抽薹期
| QTL
| [26]
|
| 水稻Rice | 籼稻 | 4亲本DC1 | 双列杂交 | 495 F6 | 6K SNP芯片 | 籽粒产量性状、穗相关性状、分蘖相关性状、抽穗期、苗期和株高
| GWAS | [23,28-31] |
| | 4亲本DC2 | 双列杂交 | 525 F6 | | | |
| | 8亲本
| Funnel杂交
| 668 F6
| 55K SNP芯片,
GBS | | |
| 籼稻 | 8亲本 | Funnel杂交 | 1328 S7 | GBS: 17387 SNP | 产量性状、稻米品质、开花期、株高、粒型、抗旱性、耐涝性、耐盐性、抗病性(稻瘟病、白叶枯病、褐斑病)
| QTL,GWAS | [32⇓-34] |
| | MAGIC PLUS | ― | 144 S8 | | | |
| | MAGIC PLUS
DH系 | ―
| 76 DH
| | | |
| 粳稻 | 8亲本 | Funnel杂交 | 500 S5 | | | |
| 籼―粳 | GLOBAL MAGIC | ― | 1402 S7 | | | |
小麦Wheat
| 春小麦
| 4亲本
| Funnel杂交
| 1579 F6
| 826 DArTs、283SNPs、53SSRs,9K SNP芯片,90K SNP芯片 | 株高、籽粒重、
胚芽鞘长度
| LMC,QTL
| [13,35-36]
|
| 冬小麦
| 8亲本
| Funnel杂交
| 1091 F7
| 90K SNP芯片
| 产量、开花期、
病害和株高 | LMC,QTL
| [37-38]
|
| 全麦粉
| 4亲本
| Funnel杂交
| 332 F6
| SSR
| 产量性状、
抗白粉病 | QTL,LMC
| [39]
|
| 欧洲
小麦 | 60亲本
| 随机杂交
| 1000 S4
| 9K SNP芯片
| 抽穗期
| LDA,GWAS
| [40]
|
| | | | | 14个KASPar SNP | | | |
大麦
Hordeum | – | 8亲本 | Funnel杂交 | 5000 DH | 9K SNP芯片 | 开花期 | QTL | [41] |
| 8亲本 | Funnel杂交 | 642 F5 | 9K SNP芯片 | 黄斑病 | LMC,QTL | [42] |
| 棉花Cotton | 陆地棉 | 11亲本 | 随机杂交 | 547 F5 | GBS | 纤维品质 | GWAS,QTL | [43] |
| | 8亲本
| 随机杂交
| 960 MLs
| SLAF-seq
| 开花期、株型、
果枝数 | GWAS,QTL
| [44]
|
| | 11亲本 | 随机杂交 | 550 RIL | 47K SNP芯片 | 株高、茎秆质量 | GWAS,QTL | [45] |
高粱
Sorghum | –
| 29亲本
| 随机杂交
| 1000 F6
| GBS
| 株高
| GWAS,QTL
| [46]
|
作物
Crop | 类型
Type | 亲本选择
Parental selection | 杂交方式
Hybridization | 群体规模
Group size | 基因型鉴定
Genotyping | 研究性状
Research trait | 分析方法
Analytical method | 参考文献
Reference |
烟草
Tobacco | –
| 8亲本
| 双列杂交
| 600 F4
| 430K 芯片
| 株型、烟叶品质
| GWAS
| [47]
|
玉米
Maize | –
| 8亲本
| Funnel杂交
| 1636 F6
| GBS
| 开花期、株高、
穗位高 | LMC,AM,QTL | [48]
|
| | 4亲本
| Funnel杂交
| 1149 S3
| 118K SNP 芯片
| 株高、穗高、散
粉期、吐丝期 | QTL,GWAS
| [49]
|
| | 8亲本
| Funnel杂交
| 401 RIL
| 56K SNP 芯片
| 苗重、苗长和
黄萎病 | QTL,NAM
| [50]
|
| | 8亲本
| Funnel杂交
| 700F6
| 110K SNP
芯片 | 镰孢菌穗腐病
| LMC,AM,QTL | [51]
|
| | 8亲本
| Funnel杂交
| 406 RIL
| 100K SNP
芯片,GBS | 发芽率、叶绿素
和出苗期 | GWAS,QTL
| [52]
|
| | 8亲本
| Funnel杂交
| 368 MLs
| 1000K SNP芯片,GBS | 籽粒含油量
| GWAS,QTL
| [53]
|
| | 16亲本
| Funnel杂交
| 513 RIL
| 500K SNP芯片
| 株高、叶形态、
开花期、穗大小 | GWAS
| [54]
|
| | 4亲本
| Funnel杂交
| 1044 F6
| 50K SNP芯片
| 产量、吐丝期、
株高 | QTL,AM
| [55]
|
油菜
Brassica napus | 甘蓝型
| 8亲本
| 随机杂交
| 680 F6
| –
| 始花期、抗病性
| SSD
| [56]
|
Fn表示构建F1的杂交过程和F1自交过程的总代数,Sn表示F1植株自交的代数,DH(doubled haploid)表示双单倍体系,GBS(genotyping by sequencing)表示基于测序的基因分型,QTL(quantitative trait locus)表示QTL定位,LDA(linkage disequilibrium analysis)表示连锁不平衡分析,LMC(linkage map construction)表示连锁图谱构建,AM(association mapping)表示关联作图,GWAS(genome wide association study)表示全基因组关联分析,SNP(single nucleotide polymorphisms)表示单核苷酸多态性,Funnel杂交表示“漏斗”型杂交,SSR(simple sequence repeat)表示简单重复序列,SSD(single seed descent)表示单粒传法,RIL(recombinant inbred lines)表示重组自交系,ML(lines)表示自交系,NAM(nested association map)表示巢式关联图谱 ...
An eight parent multiparent advanced generation inter-cross population for winter-sown wheat:creation,properties,and validation
2
2014
... Application of MAGIC group in plant genetics research
Table 1 作物
Crop | 类型
Type | 亲本选择
Parental selection | 杂交方式
Hybridization | 群体规模
Group size | 基因型鉴定
Genotyping | 研究性状
Research trait | 分析方法
Analytical method | 参考文献
Reference |
拟南芥
Arabidopsis | –
| 19亲本
| 随机杂交
| 1026 F4
| 1260 SNPs
| 抽薹期
| QTL
| [26]
|
| 水稻Rice | 籼稻 | 4亲本DC1 | 双列杂交 | 495 F6 | 6K SNP芯片 | 籽粒产量性状、穗相关性状、分蘖相关性状、抽穗期、苗期和株高
| GWAS | [23,28-31] |
| | 4亲本DC2 | 双列杂交 | 525 F6 | | | |
| | 8亲本
| Funnel杂交
| 668 F6
| 55K SNP芯片,
GBS | | |
| 籼稻 | 8亲本 | Funnel杂交 | 1328 S7 | GBS: 17387 SNP | 产量性状、稻米品质、开花期、株高、粒型、抗旱性、耐涝性、耐盐性、抗病性(稻瘟病、白叶枯病、褐斑病)
| QTL,GWAS | [32⇓-34] |
| | MAGIC PLUS | ― | 144 S8 | | | |
| | MAGIC PLUS
DH系 | ―
| 76 DH
| | | |
| 粳稻 | 8亲本 | Funnel杂交 | 500 S5 | | | |
| 籼―粳 | GLOBAL MAGIC | ― | 1402 S7 | | | |
小麦Wheat
| 春小麦
| 4亲本
| Funnel杂交
| 1579 F6
| 826 DArTs、283SNPs、53SSRs,9K SNP芯片,90K SNP芯片 | 株高、籽粒重、
胚芽鞘长度
| LMC,QTL
| [13,35-36]
|
| 冬小麦
| 8亲本
| Funnel杂交
| 1091 F7
| 90K SNP芯片
| 产量、开花期、
病害和株高 | LMC,QTL
| [37-38]
|
| 全麦粉
| 4亲本
| Funnel杂交
| 332 F6
| SSR
| 产量性状、
抗白粉病 | QTL,LMC
| [39]
|
| 欧洲
小麦 | 60亲本
| 随机杂交
| 1000 S4
| 9K SNP芯片
| 抽穗期
| LDA,GWAS
| [40]
|
| | | | | 14个KASPar SNP | | | |
大麦
Hordeum | – | 8亲本 | Funnel杂交 | 5000 DH | 9K SNP芯片 | 开花期 | QTL | [41] |
| 8亲本 | Funnel杂交 | 642 F5 | 9K SNP芯片 | 黄斑病 | LMC,QTL | [42] |
| 棉花Cotton | 陆地棉 | 11亲本 | 随机杂交 | 547 F5 | GBS | 纤维品质 | GWAS,QTL | [43] |
| | 8亲本
| 随机杂交
| 960 MLs
| SLAF-seq
| 开花期、株型、
果枝数 | GWAS,QTL
| [44]
|
| | 11亲本 | 随机杂交 | 550 RIL | 47K SNP芯片 | 株高、茎秆质量 | GWAS,QTL | [45] |
高粱
Sorghum | –
| 29亲本
| 随机杂交
| 1000 F6
| GBS
| 株高
| GWAS,QTL
| [46]
|
作物
Crop | 类型
Type | 亲本选择
Parental selection | 杂交方式
Hybridization | 群体规模
Group size | 基因型鉴定
Genotyping | 研究性状
Research trait | 分析方法
Analytical method | 参考文献
Reference |
烟草
Tobacco | –
| 8亲本
| 双列杂交
| 600 F4
| 430K 芯片
| 株型、烟叶品质
| GWAS
| [47]
|
玉米
Maize | –
| 8亲本
| Funnel杂交
| 1636 F6
| GBS
| 开花期、株高、
穗位高 | LMC,AM,QTL | [48]
|
| | 4亲本
| Funnel杂交
| 1149 S3
| 118K SNP 芯片
| 株高、穗高、散
粉期、吐丝期 | QTL,GWAS
| [49]
|
| | 8亲本
| Funnel杂交
| 401 RIL
| 56K SNP 芯片
| 苗重、苗长和
黄萎病 | QTL,NAM
| [50]
|
| | 8亲本
| Funnel杂交
| 700F6
| 110K SNP
芯片 | 镰孢菌穗腐病
| LMC,AM,QTL | [51]
|
| | 8亲本
| Funnel杂交
| 406 RIL
| 100K SNP
芯片,GBS | 发芽率、叶绿素
和出苗期 | GWAS,QTL
| [52]
|
| | 8亲本
| Funnel杂交
| 368 MLs
| 1000K SNP芯片,GBS | 籽粒含油量
| GWAS,QTL
| [53]
|
| | 16亲本
| Funnel杂交
| 513 RIL
| 500K SNP芯片
| 株高、叶形态、
开花期、穗大小 | GWAS
| [54]
|
| | 4亲本
| Funnel杂交
| 1044 F6
| 50K SNP芯片
| 产量、吐丝期、
株高 | QTL,AM
| [55]
|
油菜
Brassica napus | 甘蓝型
| 8亲本
| 随机杂交
| 680 F6
| –
| 始花期、抗病性
| SSD
| [56]
|
Fn表示构建F1的杂交过程和F1自交过程的总代数,Sn表示F1植株自交的代数,DH(doubled haploid)表示双单倍体系,GBS(genotyping by sequencing)表示基于测序的基因分型,QTL(quantitative trait locus)表示QTL定位,LDA(linkage disequilibrium analysis)表示连锁不平衡分析,LMC(linkage map construction)表示连锁图谱构建,AM(association mapping)表示关联作图,GWAS(genome wide association study)表示全基因组关联分析,SNP(single nucleotide polymorphisms)表示单核苷酸多态性,Funnel杂交表示“漏斗”型杂交,SSR(simple sequence repeat)表示简单重复序列,SSD(single seed descent)表示单粒传法,RIL(recombinant inbred lines)表示重组自交系,ML(lines)表示自交系,NAM(nested association map)表示巢式关联图谱 ...
... 小麦基因组结构复杂,对于MAGIC群体也进行了深入遗传研究.第1个小麦MAGIC群体是Huang等[35]使用4个澳洲春小麦作为亲本构建了包含1579个 RILs的MAGIC群体.利用1162个DArT(diversity arrays technology)、SNP和SSR(simple sequence repeat)标记鉴定了871个家系的基因型并且构建了遗传图谱,检测到9个株高和8个百粒重的QTL,其中有3个株高QTL和已知基因相邻.Mackay等[37,57]采用8个冬小麦亲本构建了1个包含1091个家系的MAGIC群体.通过使用Illumina平台的90 000 SNP小麦芯片鉴定720个家系和8个亲本的基因型,共获得62 543个SNP标记.他们利用SNP标记通过连锁不平衡分析了基因组重组.小麦MAGIC群体是数据收集中进展最快的群体,推动了小麦遗传分析方法的发展,无论是在连锁图谱构建领域,还是标记和性状的关联定位,都取得了很好的进展. ...
The statistical analysis of multi-environment data:modelling genotype-by-environment interaction and its genetic basis
2
2013
... Application of MAGIC group in plant genetics research
Table 1 作物
Crop | 类型
Type | 亲本选择
Parental selection | 杂交方式
Hybridization | 群体规模
Group size | 基因型鉴定
Genotyping | 研究性状
Research trait | 分析方法
Analytical method | 参考文献
Reference |
拟南芥
Arabidopsis | –
| 19亲本
| 随机杂交
| 1026 F4
| 1260 SNPs
| 抽薹期
| QTL
| [26]
|
| 水稻Rice | 籼稻 | 4亲本DC1 | 双列杂交 | 495 F6 | 6K SNP芯片 | 籽粒产量性状、穗相关性状、分蘖相关性状、抽穗期、苗期和株高
| GWAS | [23,28-31] |
| | 4亲本DC2 | 双列杂交 | 525 F6 | | | |
| | 8亲本
| Funnel杂交
| 668 F6
| 55K SNP芯片,
GBS | | |
| 籼稻 | 8亲本 | Funnel杂交 | 1328 S7 | GBS: 17387 SNP | 产量性状、稻米品质、开花期、株高、粒型、抗旱性、耐涝性、耐盐性、抗病性(稻瘟病、白叶枯病、褐斑病)
| QTL,GWAS | [32⇓-34] |
| | MAGIC PLUS | ― | 144 S8 | | | |
| | MAGIC PLUS
DH系 | ―
| 76 DH
| | | |
| 粳稻 | 8亲本 | Funnel杂交 | 500 S5 | | | |
| 籼―粳 | GLOBAL MAGIC | ― | 1402 S7 | | | |
小麦Wheat
| 春小麦
| 4亲本
| Funnel杂交
| 1579 F6
| 826 DArTs、283SNPs、53SSRs,9K SNP芯片,90K SNP芯片 | 株高、籽粒重、
胚芽鞘长度
| LMC,QTL
| [13,35-36]
|
| 冬小麦
| 8亲本
| Funnel杂交
| 1091 F7
| 90K SNP芯片
| 产量、开花期、
病害和株高 | LMC,QTL
| [37-38]
|
| 全麦粉
| 4亲本
| Funnel杂交
| 332 F6
| SSR
| 产量性状、
抗白粉病 | QTL,LMC
| [39]
|
| 欧洲
小麦 | 60亲本
| 随机杂交
| 1000 S4
| 9K SNP芯片
| 抽穗期
| LDA,GWAS
| [40]
|
| | | | | 14个KASPar SNP | | | |
大麦
Hordeum | – | 8亲本 | Funnel杂交 | 5000 DH | 9K SNP芯片 | 开花期 | QTL | [41] |
| 8亲本 | Funnel杂交 | 642 F5 | 9K SNP芯片 | 黄斑病 | LMC,QTL | [42] |
| 棉花Cotton | 陆地棉 | 11亲本 | 随机杂交 | 547 F5 | GBS | 纤维品质 | GWAS,QTL | [43] |
| | 8亲本
| 随机杂交
| 960 MLs
| SLAF-seq
| 开花期、株型、
果枝数 | GWAS,QTL
| [44]
|
| | 11亲本 | 随机杂交 | 550 RIL | 47K SNP芯片 | 株高、茎秆质量 | GWAS,QTL | [45] |
高粱
Sorghum | –
| 29亲本
| 随机杂交
| 1000 F6
| GBS
| 株高
| GWAS,QTL
| [46]
|
作物
Crop | 类型
Type | 亲本选择
Parental selection | 杂交方式
Hybridization | 群体规模
Group size | 基因型鉴定
Genotyping | 研究性状
Research trait | 分析方法
Analytical method | 参考文献
Reference |
烟草
Tobacco | –
| 8亲本
| 双列杂交
| 600 F4
| 430K 芯片
| 株型、烟叶品质
| GWAS
| [47]
|
玉米
Maize | –
| 8亲本
| Funnel杂交
| 1636 F6
| GBS
| 开花期、株高、
穗位高 | LMC,AM,QTL | [48]
|
| | 4亲本
| Funnel杂交
| 1149 S3
| 118K SNP 芯片
| 株高、穗高、散
粉期、吐丝期 | QTL,GWAS
| [49]
|
| | 8亲本
| Funnel杂交
| 401 RIL
| 56K SNP 芯片
| 苗重、苗长和
黄萎病 | QTL,NAM
| [50]
|
| | 8亲本
| Funnel杂交
| 700F6
| 110K SNP
芯片 | 镰孢菌穗腐病
| LMC,AM,QTL | [51]
|
| | 8亲本
| Funnel杂交
| 406 RIL
| 100K SNP
芯片,GBS | 发芽率、叶绿素
和出苗期 | GWAS,QTL
| [52]
|
| | 8亲本
| Funnel杂交
| 368 MLs
| 1000K SNP芯片,GBS | 籽粒含油量
| GWAS,QTL
| [53]
|
| | 16亲本
| Funnel杂交
| 513 RIL
| 500K SNP芯片
| 株高、叶形态、
开花期、穗大小 | GWAS
| [54]
|
| | 4亲本
| Funnel杂交
| 1044 F6
| 50K SNP芯片
| 产量、吐丝期、
株高 | QTL,AM
| [55]
|
油菜
Brassica napus | 甘蓝型
| 8亲本
| 随机杂交
| 680 F6
| –
| 始花期、抗病性
| SSD
| [56]
|
Fn表示构建F1的杂交过程和F1自交过程的总代数,Sn表示F1植株自交的代数,DH(doubled haploid)表示双单倍体系,GBS(genotyping by sequencing)表示基于测序的基因分型,QTL(quantitative trait locus)表示QTL定位,LDA(linkage disequilibrium analysis)表示连锁不平衡分析,LMC(linkage map construction)表示连锁图谱构建,AM(association mapping)表示关联作图,GWAS(genome wide association study)表示全基因组关联分析,SNP(single nucleotide polymorphisms)表示单核苷酸多态性,Funnel杂交表示“漏斗”型杂交,SSR(simple sequence repeat)表示简单重复序列,SSD(single seed descent)表示单粒传法,RIL(recombinant inbred lines)表示重组自交系,ML(lines)表示自交系,NAM(nested association map)表示巢式关联图谱 ...
... 对于MAGIC,多亲本全基因组QTL分析可以利用全基因组平均区间图谱(whole-genome average interval mapping,WGAIM或MPWGAIM)延伸到多变量的情况[65-66].MPWGAIM涉及到每个潜在的QTL位点和每个亲本遗传等位基因的概率,允许连锁图谱上的每个区间或每个标记上都有1个QTL,无论在间隔内还是在标记处,都可以对假设QTL大小随机效应进行显著性检验来确定QTL是否存在.而Scutari等[67]采用了另一种贝叶斯网络的方法,利用冬小麦MAGIC群体的数据来探索贝叶斯网络同时建模分析多个数量性状的框架.结果表明,这种方法相当于多元最优线性无偏预测(genetic best liear unbiased prediction,GBLUP),因为MAGIC群体非常低的群体结构和大的样本容量使预测模型具有一个理想的环境,因此,在预测性能上与单性状GBLUP具有竞争性.此外,Malosetti等[38]说他们的双亲群体多变量模型可以很容易地扩展到多亲本群体.虽然MPWGAIM和贝叶斯网络都已应用到小麦MAGIC数据,但双方重点不同,未进行直接比较.对于所有的大数据统计分析方法,由于运用了较复杂的模型,需要耗费大量的时间和计算资源. ...
高筋全麦粉用途小麦的MAGIC群体法培育及品质检测
2
2019
... Application of MAGIC group in plant genetics research
Table 1 作物
Crop | 类型
Type | 亲本选择
Parental selection | 杂交方式
Hybridization | 群体规模
Group size | 基因型鉴定
Genotyping | 研究性状
Research trait | 分析方法
Analytical method | 参考文献
Reference |
拟南芥
Arabidopsis | –
| 19亲本
| 随机杂交
| 1026 F4
| 1260 SNPs
| 抽薹期
| QTL
| [26]
|
| 水稻Rice | 籼稻 | 4亲本DC1 | 双列杂交 | 495 F6 | 6K SNP芯片 | 籽粒产量性状、穗相关性状、分蘖相关性状、抽穗期、苗期和株高
| GWAS | [23,28-31] |
| | 4亲本DC2 | 双列杂交 | 525 F6 | | | |
| | 8亲本
| Funnel杂交
| 668 F6
| 55K SNP芯片,
GBS | | |
| 籼稻 | 8亲本 | Funnel杂交 | 1328 S7 | GBS: 17387 SNP | 产量性状、稻米品质、开花期、株高、粒型、抗旱性、耐涝性、耐盐性、抗病性(稻瘟病、白叶枯病、褐斑病)
| QTL,GWAS | [32⇓-34] |
| | MAGIC PLUS | ― | 144 S8 | | | |
| | MAGIC PLUS
DH系 | ―
| 76 DH
| | | |
| 粳稻 | 8亲本 | Funnel杂交 | 500 S5 | | | |
| 籼―粳 | GLOBAL MAGIC | ― | 1402 S7 | | | |
小麦Wheat
| 春小麦
| 4亲本
| Funnel杂交
| 1579 F6
| 826 DArTs、283SNPs、53SSRs,9K SNP芯片,90K SNP芯片 | 株高、籽粒重、
胚芽鞘长度
| LMC,QTL
| [13,35-36]
|
| 冬小麦
| 8亲本
| Funnel杂交
| 1091 F7
| 90K SNP芯片
| 产量、开花期、
病害和株高 | LMC,QTL
| [37-38]
|
| 全麦粉
| 4亲本
| Funnel杂交
| 332 F6
| SSR
| 产量性状、
抗白粉病 | QTL,LMC
| [39]
|
| 欧洲
小麦 | 60亲本
| 随机杂交
| 1000 S4
| 9K SNP芯片
| 抽穗期
| LDA,GWAS
| [40]
|
| | | | | 14个KASPar SNP | | | |
大麦
Hordeum | – | 8亲本 | Funnel杂交 | 5000 DH | 9K SNP芯片 | 开花期 | QTL | [41] |
| 8亲本 | Funnel杂交 | 642 F5 | 9K SNP芯片 | 黄斑病 | LMC,QTL | [42] |
| 棉花Cotton | 陆地棉 | 11亲本 | 随机杂交 | 547 F5 | GBS | 纤维品质 | GWAS,QTL | [43] |
| | 8亲本
| 随机杂交
| 960 MLs
| SLAF-seq
| 开花期、株型、
果枝数 | GWAS,QTL
| [44]
|
| | 11亲本 | 随机杂交 | 550 RIL | 47K SNP芯片 | 株高、茎秆质量 | GWAS,QTL | [45] |
高粱
Sorghum | –
| 29亲本
| 随机杂交
| 1000 F6
| GBS
| 株高
| GWAS,QTL
| [46]
|
作物
Crop | 类型
Type | 亲本选择
Parental selection | 杂交方式
Hybridization | 群体规模
Group size | 基因型鉴定
Genotyping | 研究性状
Research trait | 分析方法
Analytical method | 参考文献
Reference |
烟草
Tobacco | –
| 8亲本
| 双列杂交
| 600 F4
| 430K 芯片
| 株型、烟叶品质
| GWAS
| [47]
|
玉米
Maize | –
| 8亲本
| Funnel杂交
| 1636 F6
| GBS
| 开花期、株高、
穗位高 | LMC,AM,QTL | [48]
|
| | 4亲本
| Funnel杂交
| 1149 S3
| 118K SNP 芯片
| 株高、穗高、散
粉期、吐丝期 | QTL,GWAS
| [49]
|
| | 8亲本
| Funnel杂交
| 401 RIL
| 56K SNP 芯片
| 苗重、苗长和
黄萎病 | QTL,NAM
| [50]
|
| | 8亲本
| Funnel杂交
| 700F6
| 110K SNP
芯片 | 镰孢菌穗腐病
| LMC,AM,QTL | [51]
|
| | 8亲本
| Funnel杂交
| 406 RIL
| 100K SNP
芯片,GBS | 发芽率、叶绿素
和出苗期 | GWAS,QTL
| [52]
|
| | 8亲本
| Funnel杂交
| 368 MLs
| 1000K SNP芯片,GBS | 籽粒含油量
| GWAS,QTL
| [53]
|
| | 16亲本
| Funnel杂交
| 513 RIL
| 500K SNP芯片
| 株高、叶形态、
开花期、穗大小 | GWAS
| [54]
|
| | 4亲本
| Funnel杂交
| 1044 F6
| 50K SNP芯片
| 产量、吐丝期、
株高 | QTL,AM
| [55]
|
油菜
Brassica napus | 甘蓝型
| 8亲本
| 随机杂交
| 680 F6
| –
| 始花期、抗病性
| SSD
| [56]
|
Fn表示构建F1的杂交过程和F1自交过程的总代数,Sn表示F1植株自交的代数,DH(doubled haploid)表示双单倍体系,GBS(genotyping by sequencing)表示基于测序的基因分型,QTL(quantitative trait locus)表示QTL定位,LDA(linkage disequilibrium analysis)表示连锁不平衡分析,LMC(linkage map construction)表示连锁图谱构建,AM(association mapping)表示关联作图,GWAS(genome wide association study)表示全基因组关联分析,SNP(single nucleotide polymorphisms)表示单核苷酸多态性,Funnel杂交表示“漏斗”型杂交,SSR(simple sequence repeat)表示简单重复序列,SSD(single seed descent)表示单粒传法,RIL(recombinant inbred lines)表示重组自交系,ML(lines)表示自交系,NAM(nested association map)表示巢式关联图谱 ...
... 纪耀勇[39]为获得适合生产高筋全麦粉和抗病性高的小麦品种,以4个来自不同育种项目的具有丰富遗传多样性的优良品系为亲本,构建了由332个独立稳定株系组成的小麦MAGIC群体,并对每个株系的小麦产量、小麦白粉病抗性及其全麦粉加工品质进行检测和鉴定,从中筛选出一些适合生产高筋全麦粉的小麦株系.同时构建了高效SSR标记库,对抗小麦白粉病基因Pm21和PmV进行精细定位,并开发可用于辅助育种的实用型分子标记.对硬度相关基因Pina和Pinb进行克隆,并对MAGIC群体株系进行Pinb基因类型鉴定.适合于生产高筋全麦粉的株系167号含有抗小麦白粉病基因PmV,为Pinb-D1b型,具有培育成高筋全麦粉用途小麦的潜在应用价值. ...
Effciently tracking selection in a multiparental population:the case of earliness in wheat
1
2015
... Application of MAGIC group in plant genetics research
Table 1 作物
Crop | 类型
Type | 亲本选择
Parental selection | 杂交方式
Hybridization | 群体规模
Group size | 基因型鉴定
Genotyping | 研究性状
Research trait | 分析方法
Analytical method | 参考文献
Reference |
拟南芥
Arabidopsis | –
| 19亲本
| 随机杂交
| 1026 F4
| 1260 SNPs
| 抽薹期
| QTL
| [26]
|
| 水稻Rice | 籼稻 | 4亲本DC1 | 双列杂交 | 495 F6 | 6K SNP芯片 | 籽粒产量性状、穗相关性状、分蘖相关性状、抽穗期、苗期和株高
| GWAS | [23,28-31] |
| | 4亲本DC2 | 双列杂交 | 525 F6 | | | |
| | 8亲本
| Funnel杂交
| 668 F6
| 55K SNP芯片,
GBS | | |
| 籼稻 | 8亲本 | Funnel杂交 | 1328 S7 | GBS: 17387 SNP | 产量性状、稻米品质、开花期、株高、粒型、抗旱性、耐涝性、耐盐性、抗病性(稻瘟病、白叶枯病、褐斑病)
| QTL,GWAS | [32⇓-34] |
| | MAGIC PLUS | ― | 144 S8 | | | |
| | MAGIC PLUS
DH系 | ―
| 76 DH
| | | |
| 粳稻 | 8亲本 | Funnel杂交 | 500 S5 | | | |
| 籼―粳 | GLOBAL MAGIC | ― | 1402 S7 | | | |
小麦Wheat
| 春小麦
| 4亲本
| Funnel杂交
| 1579 F6
| 826 DArTs、283SNPs、53SSRs,9K SNP芯片,90K SNP芯片 | 株高、籽粒重、
胚芽鞘长度
| LMC,QTL
| [13,35-36]
|
| 冬小麦
| 8亲本
| Funnel杂交
| 1091 F7
| 90K SNP芯片
| 产量、开花期、
病害和株高 | LMC,QTL
| [37-38]
|
| 全麦粉
| 4亲本
| Funnel杂交
| 332 F6
| SSR
| 产量性状、
抗白粉病 | QTL,LMC
| [39]
|
| 欧洲
小麦 | 60亲本
| 随机杂交
| 1000 S4
| 9K SNP芯片
| 抽穗期
| LDA,GWAS
| [40]
|
| | | | | 14个KASPar SNP | | | |
大麦
Hordeum | – | 8亲本 | Funnel杂交 | 5000 DH | 9K SNP芯片 | 开花期 | QTL | [41] |
| 8亲本 | Funnel杂交 | 642 F5 | 9K SNP芯片 | 黄斑病 | LMC,QTL | [42] |
| 棉花Cotton | 陆地棉 | 11亲本 | 随机杂交 | 547 F5 | GBS | 纤维品质 | GWAS,QTL | [43] |
| | 8亲本
| 随机杂交
| 960 MLs
| SLAF-seq
| 开花期、株型、
果枝数 | GWAS,QTL
| [44]
|
| | 11亲本 | 随机杂交 | 550 RIL | 47K SNP芯片 | 株高、茎秆质量 | GWAS,QTL | [45] |
高粱
Sorghum | –
| 29亲本
| 随机杂交
| 1000 F6
| GBS
| 株高
| GWAS,QTL
| [46]
|
作物
Crop | 类型
Type | 亲本选择
Parental selection | 杂交方式
Hybridization | 群体规模
Group size | 基因型鉴定
Genotyping | 研究性状
Research trait | 分析方法
Analytical method | 参考文献
Reference |
烟草
Tobacco | –
| 8亲本
| 双列杂交
| 600 F4
| 430K 芯片
| 株型、烟叶品质
| GWAS
| [47]
|
玉米
Maize | –
| 8亲本
| Funnel杂交
| 1636 F6
| GBS
| 开花期、株高、
穗位高 | LMC,AM,QTL | [48]
|
| | 4亲本
| Funnel杂交
| 1149 S3
| 118K SNP 芯片
| 株高、穗高、散
粉期、吐丝期 | QTL,GWAS
| [49]
|
| | 8亲本
| Funnel杂交
| 401 RIL
| 56K SNP 芯片
| 苗重、苗长和
黄萎病 | QTL,NAM
| [50]
|
| | 8亲本
| Funnel杂交
| 700F6
| 110K SNP
芯片 | 镰孢菌穗腐病
| LMC,AM,QTL | [51]
|
| | 8亲本
| Funnel杂交
| 406 RIL
| 100K SNP
芯片,GBS | 发芽率、叶绿素
和出苗期 | GWAS,QTL
| [52]
|
| | 8亲本
| Funnel杂交
| 368 MLs
| 1000K SNP芯片,GBS | 籽粒含油量
| GWAS,QTL
| [53]
|
| | 16亲本
| Funnel杂交
| 513 RIL
| 500K SNP芯片
| 株高、叶形态、
开花期、穗大小 | GWAS
| [54]
|
| | 4亲本
| Funnel杂交
| 1044 F6
| 50K SNP芯片
| 产量、吐丝期、
株高 | QTL,AM
| [55]
|
油菜
Brassica napus | 甘蓝型
| 8亲本
| 随机杂交
| 680 F6
| –
| 始花期、抗病性
| SSD
| [56]
|
Fn表示构建F1的杂交过程和F1自交过程的总代数,Sn表示F1植株自交的代数,DH(doubled haploid)表示双单倍体系,GBS(genotyping by sequencing)表示基于测序的基因分型,QTL(quantitative trait locus)表示QTL定位,LDA(linkage disequilibrium analysis)表示连锁不平衡分析,LMC(linkage map construction)表示连锁图谱构建,AM(association mapping)表示关联作图,GWAS(genome wide association study)表示全基因组关联分析,SNP(single nucleotide polymorphisms)表示单核苷酸多态性,Funnel杂交表示“漏斗”型杂交,SSR(simple sequence repeat)表示简单重复序列,SSD(single seed descent)表示单粒传法,RIL(recombinant inbred lines)表示重组自交系,ML(lines)表示自交系,NAM(nested association map)表示巢式关联图谱 ...
Multi-parent advanced generation intercross in barley:high-resolution quantitative trait locus mapping for flowering time as a proof of concept
2
2015
... Application of MAGIC group in plant genetics research
Table 1 作物
Crop | 类型
Type | 亲本选择
Parental selection | 杂交方式
Hybridization | 群体规模
Group size | 基因型鉴定
Genotyping | 研究性状
Research trait | 分析方法
Analytical method | 参考文献
Reference |
拟南芥
Arabidopsis | –
| 19亲本
| 随机杂交
| 1026 F4
| 1260 SNPs
| 抽薹期
| QTL
| [26]
|
| 水稻Rice | 籼稻 | 4亲本DC1 | 双列杂交 | 495 F6 | 6K SNP芯片 | 籽粒产量性状、穗相关性状、分蘖相关性状、抽穗期、苗期和株高
| GWAS | [23,28-31] |
| | 4亲本DC2 | 双列杂交 | 525 F6 | | | |
| | 8亲本
| Funnel杂交
| 668 F6
| 55K SNP芯片,
GBS | | |
| 籼稻 | 8亲本 | Funnel杂交 | 1328 S7 | GBS: 17387 SNP | 产量性状、稻米品质、开花期、株高、粒型、抗旱性、耐涝性、耐盐性、抗病性(稻瘟病、白叶枯病、褐斑病)
| QTL,GWAS | [32⇓-34] |
| | MAGIC PLUS | ― | 144 S8 | | | |
| | MAGIC PLUS
DH系 | ―
| 76 DH
| | | |
| 粳稻 | 8亲本 | Funnel杂交 | 500 S5 | | | |
| 籼―粳 | GLOBAL MAGIC | ― | 1402 S7 | | | |
小麦Wheat
| 春小麦
| 4亲本
| Funnel杂交
| 1579 F6
| 826 DArTs、283SNPs、53SSRs,9K SNP芯片,90K SNP芯片 | 株高、籽粒重、
胚芽鞘长度
| LMC,QTL
| [13,35-36]
|
| 冬小麦
| 8亲本
| Funnel杂交
| 1091 F7
| 90K SNP芯片
| 产量、开花期、
病害和株高 | LMC,QTL
| [37-38]
|
| 全麦粉
| 4亲本
| Funnel杂交
| 332 F6
| SSR
| 产量性状、
抗白粉病 | QTL,LMC
| [39]
|
| 欧洲
小麦 | 60亲本
| 随机杂交
| 1000 S4
| 9K SNP芯片
| 抽穗期
| LDA,GWAS
| [40]
|
| | | | | 14个KASPar SNP | | | |
大麦
Hordeum | – | 8亲本 | Funnel杂交 | 5000 DH | 9K SNP芯片 | 开花期 | QTL | [41] |
| 8亲本 | Funnel杂交 | 642 F5 | 9K SNP芯片 | 黄斑病 | LMC,QTL | [42] |
| 棉花Cotton | 陆地棉 | 11亲本 | 随机杂交 | 547 F5 | GBS | 纤维品质 | GWAS,QTL | [43] |
| | 8亲本
| 随机杂交
| 960 MLs
| SLAF-seq
| 开花期、株型、
果枝数 | GWAS,QTL
| [44]
|
| | 11亲本 | 随机杂交 | 550 RIL | 47K SNP芯片 | 株高、茎秆质量 | GWAS,QTL | [45] |
高粱
Sorghum | –
| 29亲本
| 随机杂交
| 1000 F6
| GBS
| 株高
| GWAS,QTL
| [46]
|
作物
Crop | 类型
Type | 亲本选择
Parental selection | 杂交方式
Hybridization | 群体规模
Group size | 基因型鉴定
Genotyping | 研究性状
Research trait | 分析方法
Analytical method | 参考文献
Reference |
烟草
Tobacco | –
| 8亲本
| 双列杂交
| 600 F4
| 430K 芯片
| 株型、烟叶品质
| GWAS
| [47]
|
玉米
Maize | –
| 8亲本
| Funnel杂交
| 1636 F6
| GBS
| 开花期、株高、
穗位高 | LMC,AM,QTL | [48]
|
| | 4亲本
| Funnel杂交
| 1149 S3
| 118K SNP 芯片
| 株高、穗高、散
粉期、吐丝期 | QTL,GWAS
| [49]
|
| | 8亲本
| Funnel杂交
| 401 RIL
| 56K SNP 芯片
| 苗重、苗长和
黄萎病 | QTL,NAM
| [50]
|
| | 8亲本
| Funnel杂交
| 700F6
| 110K SNP
芯片 | 镰孢菌穗腐病
| LMC,AM,QTL | [51]
|
| | 8亲本
| Funnel杂交
| 406 RIL
| 100K SNP
芯片,GBS | 发芽率、叶绿素
和出苗期 | GWAS,QTL
| [52]
|
| | 8亲本
| Funnel杂交
| 368 MLs
| 1000K SNP芯片,GBS | 籽粒含油量
| GWAS,QTL
| [53]
|
| | 16亲本
| Funnel杂交
| 513 RIL
| 500K SNP芯片
| 株高、叶形态、
开花期、穗大小 | GWAS
| [54]
|
| | 4亲本
| Funnel杂交
| 1044 F6
| 50K SNP芯片
| 产量、吐丝期、
株高 | QTL,AM
| [55]
|
油菜
Brassica napus | 甘蓝型
| 8亲本
| 随机杂交
| 680 F6
| –
| 始花期、抗病性
| SSD
| [56]
|
Fn表示构建F1的杂交过程和F1自交过程的总代数,Sn表示F1植株自交的代数,DH(doubled haploid)表示双单倍体系,GBS(genotyping by sequencing)表示基于测序的基因分型,QTL(quantitative trait locus)表示QTL定位,LDA(linkage disequilibrium analysis)表示连锁不平衡分析,LMC(linkage map construction)表示连锁图谱构建,AM(association mapping)表示关联作图,GWAS(genome wide association study)表示全基因组关联分析,SNP(single nucleotide polymorphisms)表示单核苷酸多态性,Funnel杂交表示“漏斗”型杂交,SSR(simple sequence repeat)表示简单重复序列,SSD(single seed descent)表示单粒传法,RIL(recombinant inbred lines)表示重组自交系,ML(lines)表示自交系,NAM(nested association map)表示巢式关联图谱 ...
... 在大麦中,Sannemann等[41]通过“漏斗”型杂交构建了1个8亲本MAGIC群体.从5000个大麦MAGIC群体的DH系中筛选出533个DH系并且用4550个标记鉴定了基因型.使用二元方法(binary approach,BA)和单倍型方法(haplotype approach,HA)扫描开花期QTL,一共检测到17个QTL.其中BA特异检测到5个,HA特异检测到2个. ...
Fine-mapping the wheat Snn1 locus conferring sensitivity to the Parastagonospora nodorum necrotrophic effector SnTox 1 using an eight founder multiparent advanced generation inter-cross population
1
2015
... Application of MAGIC group in plant genetics research
Table 1 作物
Crop | 类型
Type | 亲本选择
Parental selection | 杂交方式
Hybridization | 群体规模
Group size | 基因型鉴定
Genotyping | 研究性状
Research trait | 分析方法
Analytical method | 参考文献
Reference |
拟南芥
Arabidopsis | –
| 19亲本
| 随机杂交
| 1026 F4
| 1260 SNPs
| 抽薹期
| QTL
| [26]
|
| 水稻Rice | 籼稻 | 4亲本DC1 | 双列杂交 | 495 F6 | 6K SNP芯片 | 籽粒产量性状、穗相关性状、分蘖相关性状、抽穗期、苗期和株高
| GWAS | [23,28-31] |
| | 4亲本DC2 | 双列杂交 | 525 F6 | | | |
| | 8亲本
| Funnel杂交
| 668 F6
| 55K SNP芯片,
GBS | | |
| 籼稻 | 8亲本 | Funnel杂交 | 1328 S7 | GBS: 17387 SNP | 产量性状、稻米品质、开花期、株高、粒型、抗旱性、耐涝性、耐盐性、抗病性(稻瘟病、白叶枯病、褐斑病)
| QTL,GWAS | [32⇓-34] |
| | MAGIC PLUS | ― | 144 S8 | | | |
| | MAGIC PLUS
DH系 | ―
| 76 DH
| | | |
| 粳稻 | 8亲本 | Funnel杂交 | 500 S5 | | | |
| 籼―粳 | GLOBAL MAGIC | ― | 1402 S7 | | | |
小麦Wheat
| 春小麦
| 4亲本
| Funnel杂交
| 1579 F6
| 826 DArTs、283SNPs、53SSRs,9K SNP芯片,90K SNP芯片 | 株高、籽粒重、
胚芽鞘长度
| LMC,QTL
| [13,35-36]
|
| 冬小麦
| 8亲本
| Funnel杂交
| 1091 F7
| 90K SNP芯片
| 产量、开花期、
病害和株高 | LMC,QTL
| [37-38]
|
| 全麦粉
| 4亲本
| Funnel杂交
| 332 F6
| SSR
| 产量性状、
抗白粉病 | QTL,LMC
| [39]
|
| 欧洲
小麦 | 60亲本
| 随机杂交
| 1000 S4
| 9K SNP芯片
| 抽穗期
| LDA,GWAS
| [40]
|
| | | | | 14个KASPar SNP | | | |
大麦
Hordeum | – | 8亲本 | Funnel杂交 | 5000 DH | 9K SNP芯片 | 开花期 | QTL | [41] |
| 8亲本 | Funnel杂交 | 642 F5 | 9K SNP芯片 | 黄斑病 | LMC,QTL | [42] |
| 棉花Cotton | 陆地棉 | 11亲本 | 随机杂交 | 547 F5 | GBS | 纤维品质 | GWAS,QTL | [43] |
| | 8亲本
| 随机杂交
| 960 MLs
| SLAF-seq
| 开花期、株型、
果枝数 | GWAS,QTL
| [44]
|
| | 11亲本 | 随机杂交 | 550 RIL | 47K SNP芯片 | 株高、茎秆质量 | GWAS,QTL | [45] |
高粱
Sorghum | –
| 29亲本
| 随机杂交
| 1000 F6
| GBS
| 株高
| GWAS,QTL
| [46]
|
作物
Crop | 类型
Type | 亲本选择
Parental selection | 杂交方式
Hybridization | 群体规模
Group size | 基因型鉴定
Genotyping | 研究性状
Research trait | 分析方法
Analytical method | 参考文献
Reference |
烟草
Tobacco | –
| 8亲本
| 双列杂交
| 600 F4
| 430K 芯片
| 株型、烟叶品质
| GWAS
| [47]
|
玉米
Maize | –
| 8亲本
| Funnel杂交
| 1636 F6
| GBS
| 开花期、株高、
穗位高 | LMC,AM,QTL | [48]
|
| | 4亲本
| Funnel杂交
| 1149 S3
| 118K SNP 芯片
| 株高、穗高、散
粉期、吐丝期 | QTL,GWAS
| [49]
|
| | 8亲本
| Funnel杂交
| 401 RIL
| 56K SNP 芯片
| 苗重、苗长和
黄萎病 | QTL,NAM
| [50]
|
| | 8亲本
| Funnel杂交
| 700F6
| 110K SNP
芯片 | 镰孢菌穗腐病
| LMC,AM,QTL | [51]
|
| | 8亲本
| Funnel杂交
| 406 RIL
| 100K SNP
芯片,GBS | 发芽率、叶绿素
和出苗期 | GWAS,QTL
| [52]
|
| | 8亲本
| Funnel杂交
| 368 MLs
| 1000K SNP芯片,GBS | 籽粒含油量
| GWAS,QTL
| [53]
|
| | 16亲本
| Funnel杂交
| 513 RIL
| 500K SNP芯片
| 株高、叶形态、
开花期、穗大小 | GWAS
| [54]
|
| | 4亲本
| Funnel杂交
| 1044 F6
| 50K SNP芯片
| 产量、吐丝期、
株高 | QTL,AM
| [55]
|
油菜
Brassica napus | 甘蓝型
| 8亲本
| 随机杂交
| 680 F6
| –
| 始花期、抗病性
| SSD
| [56]
|
Fn表示构建F1的杂交过程和F1自交过程的总代数,Sn表示F1植株自交的代数,DH(doubled haploid)表示双单倍体系,GBS(genotyping by sequencing)表示基于测序的基因分型,QTL(quantitative trait locus)表示QTL定位,LDA(linkage disequilibrium analysis)表示连锁不平衡分析,LMC(linkage map construction)表示连锁图谱构建,AM(association mapping)表示关联作图,GWAS(genome wide association study)表示全基因组关联分析,SNP(single nucleotide polymorphisms)表示单核苷酸多态性,Funnel杂交表示“漏斗”型杂交,SSR(simple sequence repeat)表示简单重复序列,SSD(single seed descent)表示单粒传法,RIL(recombinant inbred lines)表示重组自交系,ML(lines)表示自交系,NAM(nested association map)表示巢式关联图谱 ...
A MAGIC population-based genome-wide association study reveals functional association of GhRBB1_A07 gene with superior fiber quality in cotton
2
2016
... Application of MAGIC group in plant genetics research
Table 1 作物
Crop | 类型
Type | 亲本选择
Parental selection | 杂交方式
Hybridization | 群体规模
Group size | 基因型鉴定
Genotyping | 研究性状
Research trait | 分析方法
Analytical method | 参考文献
Reference |
拟南芥
Arabidopsis | –
| 19亲本
| 随机杂交
| 1026 F4
| 1260 SNPs
| 抽薹期
| QTL
| [26]
|
| 水稻Rice | 籼稻 | 4亲本DC1 | 双列杂交 | 495 F6 | 6K SNP芯片 | 籽粒产量性状、穗相关性状、分蘖相关性状、抽穗期、苗期和株高
| GWAS | [23,28-31] |
| | 4亲本DC2 | 双列杂交 | 525 F6 | | | |
| | 8亲本
| Funnel杂交
| 668 F6
| 55K SNP芯片,
GBS | | |
| 籼稻 | 8亲本 | Funnel杂交 | 1328 S7 | GBS: 17387 SNP | 产量性状、稻米品质、开花期、株高、粒型、抗旱性、耐涝性、耐盐性、抗病性(稻瘟病、白叶枯病、褐斑病)
| QTL,GWAS | [32⇓-34] |
| | MAGIC PLUS | ― | 144 S8 | | | |
| | MAGIC PLUS
DH系 | ―
| 76 DH
| | | |
| 粳稻 | 8亲本 | Funnel杂交 | 500 S5 | | | |
| 籼―粳 | GLOBAL MAGIC | ― | 1402 S7 | | | |
小麦Wheat
| 春小麦
| 4亲本
| Funnel杂交
| 1579 F6
| 826 DArTs、283SNPs、53SSRs,9K SNP芯片,90K SNP芯片 | 株高、籽粒重、
胚芽鞘长度
| LMC,QTL
| [13,35-36]
|
| 冬小麦
| 8亲本
| Funnel杂交
| 1091 F7
| 90K SNP芯片
| 产量、开花期、
病害和株高 | LMC,QTL
| [37-38]
|
| 全麦粉
| 4亲本
| Funnel杂交
| 332 F6
| SSR
| 产量性状、
抗白粉病 | QTL,LMC
| [39]
|
| 欧洲
小麦 | 60亲本
| 随机杂交
| 1000 S4
| 9K SNP芯片
| 抽穗期
| LDA,GWAS
| [40]
|
| | | | | 14个KASPar SNP | | | |
大麦
Hordeum | – | 8亲本 | Funnel杂交 | 5000 DH | 9K SNP芯片 | 开花期 | QTL | [41] |
| 8亲本 | Funnel杂交 | 642 F5 | 9K SNP芯片 | 黄斑病 | LMC,QTL | [42] |
| 棉花Cotton | 陆地棉 | 11亲本 | 随机杂交 | 547 F5 | GBS | 纤维品质 | GWAS,QTL | [43] |
| | 8亲本
| 随机杂交
| 960 MLs
| SLAF-seq
| 开花期、株型、
果枝数 | GWAS,QTL
| [44]
|
| | 11亲本 | 随机杂交 | 550 RIL | 47K SNP芯片 | 株高、茎秆质量 | GWAS,QTL | [45] |
高粱
Sorghum | –
| 29亲本
| 随机杂交
| 1000 F6
| GBS
| 株高
| GWAS,QTL
| [46]
|
作物
Crop | 类型
Type | 亲本选择
Parental selection | 杂交方式
Hybridization | 群体规模
Group size | 基因型鉴定
Genotyping | 研究性状
Research trait | 分析方法
Analytical method | 参考文献
Reference |
烟草
Tobacco | –
| 8亲本
| 双列杂交
| 600 F4
| 430K 芯片
| 株型、烟叶品质
| GWAS
| [47]
|
玉米
Maize | –
| 8亲本
| Funnel杂交
| 1636 F6
| GBS
| 开花期、株高、
穗位高 | LMC,AM,QTL | [48]
|
| | 4亲本
| Funnel杂交
| 1149 S3
| 118K SNP 芯片
| 株高、穗高、散
粉期、吐丝期 | QTL,GWAS
| [49]
|
| | 8亲本
| Funnel杂交
| 401 RIL
| 56K SNP 芯片
| 苗重、苗长和
黄萎病 | QTL,NAM
| [50]
|
| | 8亲本
| Funnel杂交
| 700F6
| 110K SNP
芯片 | 镰孢菌穗腐病
| LMC,AM,QTL | [51]
|
| | 8亲本
| Funnel杂交
| 406 RIL
| 100K SNP
芯片,GBS | 发芽率、叶绿素
和出苗期 | GWAS,QTL
| [52]
|
| | 8亲本
| Funnel杂交
| 368 MLs
| 1000K SNP芯片,GBS | 籽粒含油量
| GWAS,QTL
| [53]
|
| | 16亲本
| Funnel杂交
| 513 RIL
| 500K SNP芯片
| 株高、叶形态、
开花期、穗大小 | GWAS
| [54]
|
| | 4亲本
| Funnel杂交
| 1044 F6
| 50K SNP芯片
| 产量、吐丝期、
株高 | QTL,AM
| [55]
|
油菜
Brassica napus | 甘蓝型
| 8亲本
| 随机杂交
| 680 F6
| –
| 始花期、抗病性
| SSD
| [56]
|
Fn表示构建F1的杂交过程和F1自交过程的总代数,Sn表示F1植株自交的代数,DH(doubled haploid)表示双单倍体系,GBS(genotyping by sequencing)表示基于测序的基因分型,QTL(quantitative trait locus)表示QTL定位,LDA(linkage disequilibrium analysis)表示连锁不平衡分析,LMC(linkage map construction)表示连锁图谱构建,AM(association mapping)表示关联作图,GWAS(genome wide association study)表示全基因组关联分析,SNP(single nucleotide polymorphisms)表示单核苷酸多态性,Funnel杂交表示“漏斗”型杂交,SSR(simple sequence repeat)表示简单重复序列,SSD(single seed descent)表示单粒传法,RIL(recombinant inbred lines)表示重组自交系,ML(lines)表示自交系,NAM(nested association map)表示巢式关联图谱 ...
... 在棉花遗传学研究中,Islam等[43]和郭志军等[58]发现陆地棉MAGIC群体通过11个不同品种随机交配5代陆地棉MAGIC群体,利用6071个SNP标记,鉴定到一个影响纤维质量的基因GhRBB1_A07.有的研究[44,59-60]为了探索陆地棉重要农艺性状的遗传基础,从960 MLs的8亲本MAGIC群体中选择1个包含372个株系的MAGIC群体子集.利用60 495个SNP和表型数据进行全基因组关联作图,鉴定出177个 SNP在多个环境下与9个农艺性状显著相关.在117个QTL中有3个QTL在多种环境中是稳定的,11个QTL被证明与多个性状相关联. ...
Genome-wide association mapping for agronomic traits in an 8-way Upland cotton MAGIC population by SLAF-seq
2
2021
... Application of MAGIC group in plant genetics research
Table 1 作物
Crop | 类型
Type | 亲本选择
Parental selection | 杂交方式
Hybridization | 群体规模
Group size | 基因型鉴定
Genotyping | 研究性状
Research trait | 分析方法
Analytical method | 参考文献
Reference |
拟南芥
Arabidopsis | –
| 19亲本
| 随机杂交
| 1026 F4
| 1260 SNPs
| 抽薹期
| QTL
| [26]
|
| 水稻Rice | 籼稻 | 4亲本DC1 | 双列杂交 | 495 F6 | 6K SNP芯片 | 籽粒产量性状、穗相关性状、分蘖相关性状、抽穗期、苗期和株高
| GWAS | [23,28-31] |
| | 4亲本DC2 | 双列杂交 | 525 F6 | | | |
| | 8亲本
| Funnel杂交
| 668 F6
| 55K SNP芯片,
GBS | | |
| 籼稻 | 8亲本 | Funnel杂交 | 1328 S7 | GBS: 17387 SNP | 产量性状、稻米品质、开花期、株高、粒型、抗旱性、耐涝性、耐盐性、抗病性(稻瘟病、白叶枯病、褐斑病)
| QTL,GWAS | [32⇓-34] |
| | MAGIC PLUS | ― | 144 S8 | | | |
| | MAGIC PLUS
DH系 | ―
| 76 DH
| | | |
| 粳稻 | 8亲本 | Funnel杂交 | 500 S5 | | | |
| 籼―粳 | GLOBAL MAGIC | ― | 1402 S7 | | | |
小麦Wheat
| 春小麦
| 4亲本
| Funnel杂交
| 1579 F6
| 826 DArTs、283SNPs、53SSRs,9K SNP芯片,90K SNP芯片 | 株高、籽粒重、
胚芽鞘长度
| LMC,QTL
| [13,35-36]
|
| 冬小麦
| 8亲本
| Funnel杂交
| 1091 F7
| 90K SNP芯片
| 产量、开花期、
病害和株高 | LMC,QTL
| [37-38]
|
| 全麦粉
| 4亲本
| Funnel杂交
| 332 F6
| SSR
| 产量性状、
抗白粉病 | QTL,LMC
| [39]
|
| 欧洲
小麦 | 60亲本
| 随机杂交
| 1000 S4
| 9K SNP芯片
| 抽穗期
| LDA,GWAS
| [40]
|
| | | | | 14个KASPar SNP | | | |
大麦
Hordeum | – | 8亲本 | Funnel杂交 | 5000 DH | 9K SNP芯片 | 开花期 | QTL | [41] |
| 8亲本 | Funnel杂交 | 642 F5 | 9K SNP芯片 | 黄斑病 | LMC,QTL | [42] |
| 棉花Cotton | 陆地棉 | 11亲本 | 随机杂交 | 547 F5 | GBS | 纤维品质 | GWAS,QTL | [43] |
| | 8亲本
| 随机杂交
| 960 MLs
| SLAF-seq
| 开花期、株型、
果枝数 | GWAS,QTL
| [44]
|
| | 11亲本 | 随机杂交 | 550 RIL | 47K SNP芯片 | 株高、茎秆质量 | GWAS,QTL | [45] |
高粱
Sorghum | –
| 29亲本
| 随机杂交
| 1000 F6
| GBS
| 株高
| GWAS,QTL
| [46]
|
作物
Crop | 类型
Type | 亲本选择
Parental selection | 杂交方式
Hybridization | 群体规模
Group size | 基因型鉴定
Genotyping | 研究性状
Research trait | 分析方法
Analytical method | 参考文献
Reference |
烟草
Tobacco | –
| 8亲本
| 双列杂交
| 600 F4
| 430K 芯片
| 株型、烟叶品质
| GWAS
| [47]
|
玉米
Maize | –
| 8亲本
| Funnel杂交
| 1636 F6
| GBS
| 开花期、株高、
穗位高 | LMC,AM,QTL | [48]
|
| | 4亲本
| Funnel杂交
| 1149 S3
| 118K SNP 芯片
| 株高、穗高、散
粉期、吐丝期 | QTL,GWAS
| [49]
|
| | 8亲本
| Funnel杂交
| 401 RIL
| 56K SNP 芯片
| 苗重、苗长和
黄萎病 | QTL,NAM
| [50]
|
| | 8亲本
| Funnel杂交
| 700F6
| 110K SNP
芯片 | 镰孢菌穗腐病
| LMC,AM,QTL | [51]
|
| | 8亲本
| Funnel杂交
| 406 RIL
| 100K SNP
芯片,GBS | 发芽率、叶绿素
和出苗期 | GWAS,QTL
| [52]
|
| | 8亲本
| Funnel杂交
| 368 MLs
| 1000K SNP芯片,GBS | 籽粒含油量
| GWAS,QTL
| [53]
|
| | 16亲本
| Funnel杂交
| 513 RIL
| 500K SNP芯片
| 株高、叶形态、
开花期、穗大小 | GWAS
| [54]
|
| | 4亲本
| Funnel杂交
| 1044 F6
| 50K SNP芯片
| 产量、吐丝期、
株高 | QTL,AM
| [55]
|
油菜
Brassica napus | 甘蓝型
| 8亲本
| 随机杂交
| 680 F6
| –
| 始花期、抗病性
| SSD
| [56]
|
Fn表示构建F1的杂交过程和F1自交过程的总代数,Sn表示F1植株自交的代数,DH(doubled haploid)表示双单倍体系,GBS(genotyping by sequencing)表示基于测序的基因分型,QTL(quantitative trait locus)表示QTL定位,LDA(linkage disequilibrium analysis)表示连锁不平衡分析,LMC(linkage map construction)表示连锁图谱构建,AM(association mapping)表示关联作图,GWAS(genome wide association study)表示全基因组关联分析,SNP(single nucleotide polymorphisms)表示单核苷酸多态性,Funnel杂交表示“漏斗”型杂交,SSR(simple sequence repeat)表示简单重复序列,SSD(single seed descent)表示单粒传法,RIL(recombinant inbred lines)表示重组自交系,ML(lines)表示自交系,NAM(nested association map)表示巢式关联图谱 ...
... 在棉花遗传学研究中,Islam等[43]和郭志军等[58]发现陆地棉MAGIC群体通过11个不同品种随机交配5代陆地棉MAGIC群体,利用6071个SNP标记,鉴定到一个影响纤维质量的基因GhRBB1_A07.有的研究[44,59-60]为了探索陆地棉重要农艺性状的遗传基础,从960 MLs的8亲本MAGIC群体中选择1个包含372个株系的MAGIC群体子集.利用60 495个SNP和表型数据进行全基因组关联作图,鉴定出177个 SNP在多个环境下与9个农艺性状显著相关.在117个QTL中有3个QTL在多种环境中是稳定的,11个QTL被证明与多个性状相关联. ...
GWAS reveals consistent QTL for drought and salt tolerance in a MAGIC population of 550 lines derived from intermating of 11 Upland cotton (Gossypium hirsutum) parents
1
2020
... Application of MAGIC group in plant genetics research
Table 1 作物
Crop | 类型
Type | 亲本选择
Parental selection | 杂交方式
Hybridization | 群体规模
Group size | 基因型鉴定
Genotyping | 研究性状
Research trait | 分析方法
Analytical method | 参考文献
Reference |
拟南芥
Arabidopsis | –
| 19亲本
| 随机杂交
| 1026 F4
| 1260 SNPs
| 抽薹期
| QTL
| [26]
|
| 水稻Rice | 籼稻 | 4亲本DC1 | 双列杂交 | 495 F6 | 6K SNP芯片 | 籽粒产量性状、穗相关性状、分蘖相关性状、抽穗期、苗期和株高
| GWAS | [23,28-31] |
| | 4亲本DC2 | 双列杂交 | 525 F6 | | | |
| | 8亲本
| Funnel杂交
| 668 F6
| 55K SNP芯片,
GBS | | |
| 籼稻 | 8亲本 | Funnel杂交 | 1328 S7 | GBS: 17387 SNP | 产量性状、稻米品质、开花期、株高、粒型、抗旱性、耐涝性、耐盐性、抗病性(稻瘟病、白叶枯病、褐斑病)
| QTL,GWAS | [32⇓-34] |
| | MAGIC PLUS | ― | 144 S8 | | | |
| | MAGIC PLUS
DH系 | ―
| 76 DH
| | | |
| 粳稻 | 8亲本 | Funnel杂交 | 500 S5 | | | |
| 籼―粳 | GLOBAL MAGIC | ― | 1402 S7 | | | |
小麦Wheat
| 春小麦
| 4亲本
| Funnel杂交
| 1579 F6
| 826 DArTs、283SNPs、53SSRs,9K SNP芯片,90K SNP芯片 | 株高、籽粒重、
胚芽鞘长度
| LMC,QTL
| [13,35-36]
|
| 冬小麦
| 8亲本
| Funnel杂交
| 1091 F7
| 90K SNP芯片
| 产量、开花期、
病害和株高 | LMC,QTL
| [37-38]
|
| 全麦粉
| 4亲本
| Funnel杂交
| 332 F6
| SSR
| 产量性状、
抗白粉病 | QTL,LMC
| [39]
|
| 欧洲
小麦 | 60亲本
| 随机杂交
| 1000 S4
| 9K SNP芯片
| 抽穗期
| LDA,GWAS
| [40]
|
| | | | | 14个KASPar SNP | | | |
大麦
Hordeum | – | 8亲本 | Funnel杂交 | 5000 DH | 9K SNP芯片 | 开花期 | QTL | [41] |
| 8亲本 | Funnel杂交 | 642 F5 | 9K SNP芯片 | 黄斑病 | LMC,QTL | [42] |
| 棉花Cotton | 陆地棉 | 11亲本 | 随机杂交 | 547 F5 | GBS | 纤维品质 | GWAS,QTL | [43] |
| | 8亲本
| 随机杂交
| 960 MLs
| SLAF-seq
| 开花期、株型、
果枝数 | GWAS,QTL
| [44]
|
| | 11亲本 | 随机杂交 | 550 RIL | 47K SNP芯片 | 株高、茎秆质量 | GWAS,QTL | [45] |
高粱
Sorghum | –
| 29亲本
| 随机杂交
| 1000 F6
| GBS
| 株高
| GWAS,QTL
| [46]
|
作物
Crop | 类型
Type | 亲本选择
Parental selection | 杂交方式
Hybridization | 群体规模
Group size | 基因型鉴定
Genotyping | 研究性状
Research trait | 分析方法
Analytical method | 参考文献
Reference |
烟草
Tobacco | –
| 8亲本
| 双列杂交
| 600 F4
| 430K 芯片
| 株型、烟叶品质
| GWAS
| [47]
|
玉米
Maize | –
| 8亲本
| Funnel杂交
| 1636 F6
| GBS
| 开花期、株高、
穗位高 | LMC,AM,QTL | [48]
|
| | 4亲本
| Funnel杂交
| 1149 S3
| 118K SNP 芯片
| 株高、穗高、散
粉期、吐丝期 | QTL,GWAS
| [49]
|
| | 8亲本
| Funnel杂交
| 401 RIL
| 56K SNP 芯片
| 苗重、苗长和
黄萎病 | QTL,NAM
| [50]
|
| | 8亲本
| Funnel杂交
| 700F6
| 110K SNP
芯片 | 镰孢菌穗腐病
| LMC,AM,QTL | [51]
|
| | 8亲本
| Funnel杂交
| 406 RIL
| 100K SNP
芯片,GBS | 发芽率、叶绿素
和出苗期 | GWAS,QTL
| [52]
|
| | 8亲本
| Funnel杂交
| 368 MLs
| 1000K SNP芯片,GBS | 籽粒含油量
| GWAS,QTL
| [53]
|
| | 16亲本
| Funnel杂交
| 513 RIL
| 500K SNP芯片
| 株高、叶形态、
开花期、穗大小 | GWAS
| [54]
|
| | 4亲本
| Funnel杂交
| 1044 F6
| 50K SNP芯片
| 产量、吐丝期、
株高 | QTL,AM
| [55]
|
油菜
Brassica napus | 甘蓝型
| 8亲本
| 随机杂交
| 680 F6
| –
| 始花期、抗病性
| SSD
| [56]
|
Fn表示构建F1的杂交过程和F1自交过程的总代数,Sn表示F1植株自交的代数,DH(doubled haploid)表示双单倍体系,GBS(genotyping by sequencing)表示基于测序的基因分型,QTL(quantitative trait locus)表示QTL定位,LDA(linkage disequilibrium analysis)表示连锁不平衡分析,LMC(linkage map construction)表示连锁图谱构建,AM(association mapping)表示关联作图,GWAS(genome wide association study)表示全基因组关联分析,SNP(single nucleotide polymorphisms)表示单核苷酸多态性,Funnel杂交表示“漏斗”型杂交,SSR(simple sequence repeat)表示简单重复序列,SSD(single seed descent)表示单粒传法,RIL(recombinant inbred lines)表示重组自交系,ML(lines)表示自交系,NAM(nested association map)表示巢式关联图谱 ...
Mating design and genetic structure of a multi-parent advanced generation intercross (MAGIC) population of Sorghum (Sorghum bicolor (L.) Moench)
2
2018
... Application of MAGIC group in plant genetics research
Table 1 作物
Crop | 类型
Type | 亲本选择
Parental selection | 杂交方式
Hybridization | 群体规模
Group size | 基因型鉴定
Genotyping | 研究性状
Research trait | 分析方法
Analytical method | 参考文献
Reference |
拟南芥
Arabidopsis | –
| 19亲本
| 随机杂交
| 1026 F4
| 1260 SNPs
| 抽薹期
| QTL
| [26]
|
| 水稻Rice | 籼稻 | 4亲本DC1 | 双列杂交 | 495 F6 | 6K SNP芯片 | 籽粒产量性状、穗相关性状、分蘖相关性状、抽穗期、苗期和株高
| GWAS | [23,28-31] |
| | 4亲本DC2 | 双列杂交 | 525 F6 | | | |
| | 8亲本
| Funnel杂交
| 668 F6
| 55K SNP芯片,
GBS | | |
| 籼稻 | 8亲本 | Funnel杂交 | 1328 S7 | GBS: 17387 SNP | 产量性状、稻米品质、开花期、株高、粒型、抗旱性、耐涝性、耐盐性、抗病性(稻瘟病、白叶枯病、褐斑病)
| QTL,GWAS | [32⇓-34] |
| | MAGIC PLUS | ― | 144 S8 | | | |
| | MAGIC PLUS
DH系 | ―
| 76 DH
| | | |
| 粳稻 | 8亲本 | Funnel杂交 | 500 S5 | | | |
| 籼―粳 | GLOBAL MAGIC | ― | 1402 S7 | | | |
小麦Wheat
| 春小麦
| 4亲本
| Funnel杂交
| 1579 F6
| 826 DArTs、283SNPs、53SSRs,9K SNP芯片,90K SNP芯片 | 株高、籽粒重、
胚芽鞘长度
| LMC,QTL
| [13,35-36]
|
| 冬小麦
| 8亲本
| Funnel杂交
| 1091 F7
| 90K SNP芯片
| 产量、开花期、
病害和株高 | LMC,QTL
| [37-38]
|
| 全麦粉
| 4亲本
| Funnel杂交
| 332 F6
| SSR
| 产量性状、
抗白粉病 | QTL,LMC
| [39]
|
| 欧洲
小麦 | 60亲本
| 随机杂交
| 1000 S4
| 9K SNP芯片
| 抽穗期
| LDA,GWAS
| [40]
|
| | | | | 14个KASPar SNP | | | |
大麦
Hordeum | – | 8亲本 | Funnel杂交 | 5000 DH | 9K SNP芯片 | 开花期 | QTL | [41] |
| 8亲本 | Funnel杂交 | 642 F5 | 9K SNP芯片 | 黄斑病 | LMC,QTL | [42] |
| 棉花Cotton | 陆地棉 | 11亲本 | 随机杂交 | 547 F5 | GBS | 纤维品质 | GWAS,QTL | [43] |
| | 8亲本
| 随机杂交
| 960 MLs
| SLAF-seq
| 开花期、株型、
果枝数 | GWAS,QTL
| [44]
|
| | 11亲本 | 随机杂交 | 550 RIL | 47K SNP芯片 | 株高、茎秆质量 | GWAS,QTL | [45] |
高粱
Sorghum | –
| 29亲本
| 随机杂交
| 1000 F6
| GBS
| 株高
| GWAS,QTL
| [46]
|
作物
Crop | 类型
Type | 亲本选择
Parental selection | 杂交方式
Hybridization | 群体规模
Group size | 基因型鉴定
Genotyping | 研究性状
Research trait | 分析方法
Analytical method | 参考文献
Reference |
烟草
Tobacco | –
| 8亲本
| 双列杂交
| 600 F4
| 430K 芯片
| 株型、烟叶品质
| GWAS
| [47]
|
玉米
Maize | –
| 8亲本
| Funnel杂交
| 1636 F6
| GBS
| 开花期、株高、
穗位高 | LMC,AM,QTL | [48]
|
| | 4亲本
| Funnel杂交
| 1149 S3
| 118K SNP 芯片
| 株高、穗高、散
粉期、吐丝期 | QTL,GWAS
| [49]
|
| | 8亲本
| Funnel杂交
| 401 RIL
| 56K SNP 芯片
| 苗重、苗长和
黄萎病 | QTL,NAM
| [50]
|
| | 8亲本
| Funnel杂交
| 700F6
| 110K SNP
芯片 | 镰孢菌穗腐病
| LMC,AM,QTL | [51]
|
| | 8亲本
| Funnel杂交
| 406 RIL
| 100K SNP
芯片,GBS | 发芽率、叶绿素
和出苗期 | GWAS,QTL
| [52]
|
| | 8亲本
| Funnel杂交
| 368 MLs
| 1000K SNP芯片,GBS | 籽粒含油量
| GWAS,QTL
| [53]
|
| | 16亲本
| Funnel杂交
| 513 RIL
| 500K SNP芯片
| 株高、叶形态、
开花期、穗大小 | GWAS
| [54]
|
| | 4亲本
| Funnel杂交
| 1044 F6
| 50K SNP芯片
| 产量、吐丝期、
株高 | QTL,AM
| [55]
|
油菜
Brassica napus | 甘蓝型
| 8亲本
| 随机杂交
| 680 F6
| –
| 始花期、抗病性
| SSD
| [56]
|
Fn表示构建F1的杂交过程和F1自交过程的总代数,Sn表示F1植株自交的代数,DH(doubled haploid)表示双单倍体系,GBS(genotyping by sequencing)表示基于测序的基因分型,QTL(quantitative trait locus)表示QTL定位,LDA(linkage disequilibrium analysis)表示连锁不平衡分析,LMC(linkage map construction)表示连锁图谱构建,AM(association mapping)表示关联作图,GWAS(genome wide association study)表示全基因组关联分析,SNP(single nucleotide polymorphisms)表示单核苷酸多态性,Funnel杂交表示“漏斗”型杂交,SSR(simple sequence repeat)表示简单重复序列,SSD(single seed descent)表示单粒传法,RIL(recombinant inbred lines)表示重组自交系,ML(lines)表示自交系,NAM(nested association map)表示巢式关联图谱 ...
... Ongom等[46]利用29个亲本的高粱MAGIC群体进行全基因组关联分析,定位到了2个已知的株高基因DWARFI和DWARF3,还检测到一个位于6号染色体上新的株高QTL.刘洪泰[47]利用8个烟草特色种质为亲本构建了一个包含600个株系并且具有丰富多样性的烟草MAGIC群体,充分发挥MAGIC群体高效、高精度和低假阳性的优势,在后代群体选育出株系株型良好、抗病且烟叶品质高的材料,作为育种中间材料或直接选育新品种. ...
2
2020
... Application of MAGIC group in plant genetics research
Table 1 作物
Crop | 类型
Type | 亲本选择
Parental selection | 杂交方式
Hybridization | 群体规模
Group size | 基因型鉴定
Genotyping | 研究性状
Research trait | 分析方法
Analytical method | 参考文献
Reference |
拟南芥
Arabidopsis | –
| 19亲本
| 随机杂交
| 1026 F4
| 1260 SNPs
| 抽薹期
| QTL
| [26]
|
| 水稻Rice | 籼稻 | 4亲本DC1 | 双列杂交 | 495 F6 | 6K SNP芯片 | 籽粒产量性状、穗相关性状、分蘖相关性状、抽穗期、苗期和株高
| GWAS | [23,28-31] |
| | 4亲本DC2 | 双列杂交 | 525 F6 | | | |
| | 8亲本
| Funnel杂交
| 668 F6
| 55K SNP芯片,
GBS | | |
| 籼稻 | 8亲本 | Funnel杂交 | 1328 S7 | GBS: 17387 SNP | 产量性状、稻米品质、开花期、株高、粒型、抗旱性、耐涝性、耐盐性、抗病性(稻瘟病、白叶枯病、褐斑病)
| QTL,GWAS | [32⇓-34] |
| | MAGIC PLUS | ― | 144 S8 | | | |
| | MAGIC PLUS
DH系 | ―
| 76 DH
| | | |
| 粳稻 | 8亲本 | Funnel杂交 | 500 S5 | | | |
| 籼―粳 | GLOBAL MAGIC | ― | 1402 S7 | | | |
小麦Wheat
| 春小麦
| 4亲本
| Funnel杂交
| 1579 F6
| 826 DArTs、283SNPs、53SSRs,9K SNP芯片,90K SNP芯片 | 株高、籽粒重、
胚芽鞘长度
| LMC,QTL
| [13,35-36]
|
| 冬小麦
| 8亲本
| Funnel杂交
| 1091 F7
| 90K SNP芯片
| 产量、开花期、
病害和株高 | LMC,QTL
| [37-38]
|
| 全麦粉
| 4亲本
| Funnel杂交
| 332 F6
| SSR
| 产量性状、
抗白粉病 | QTL,LMC
| [39]
|
| 欧洲
小麦 | 60亲本
| 随机杂交
| 1000 S4
| 9K SNP芯片
| 抽穗期
| LDA,GWAS
| [40]
|
| | | | | 14个KASPar SNP | | | |
大麦
Hordeum | – | 8亲本 | Funnel杂交 | 5000 DH | 9K SNP芯片 | 开花期 | QTL | [41] |
| 8亲本 | Funnel杂交 | 642 F5 | 9K SNP芯片 | 黄斑病 | LMC,QTL | [42] |
| 棉花Cotton | 陆地棉 | 11亲本 | 随机杂交 | 547 F5 | GBS | 纤维品质 | GWAS,QTL | [43] |
| | 8亲本
| 随机杂交
| 960 MLs
| SLAF-seq
| 开花期、株型、
果枝数 | GWAS,QTL
| [44]
|
| | 11亲本 | 随机杂交 | 550 RIL | 47K SNP芯片 | 株高、茎秆质量 | GWAS,QTL | [45] |
高粱
Sorghum | –
| 29亲本
| 随机杂交
| 1000 F6
| GBS
| 株高
| GWAS,QTL
| [46]
|
作物
Crop | 类型
Type | 亲本选择
Parental selection | 杂交方式
Hybridization | 群体规模
Group size | 基因型鉴定
Genotyping | 研究性状
Research trait | 分析方法
Analytical method | 参考文献
Reference |
烟草
Tobacco | –
| 8亲本
| 双列杂交
| 600 F4
| 430K 芯片
| 株型、烟叶品质
| GWAS
| [47]
|
玉米
Maize | –
| 8亲本
| Funnel杂交
| 1636 F6
| GBS
| 开花期、株高、
穗位高 | LMC,AM,QTL | [48]
|
| | 4亲本
| Funnel杂交
| 1149 S3
| 118K SNP 芯片
| 株高、穗高、散
粉期、吐丝期 | QTL,GWAS
| [49]
|
| | 8亲本
| Funnel杂交
| 401 RIL
| 56K SNP 芯片
| 苗重、苗长和
黄萎病 | QTL,NAM
| [50]
|
| | 8亲本
| Funnel杂交
| 700F6
| 110K SNP
芯片 | 镰孢菌穗腐病
| LMC,AM,QTL | [51]
|
| | 8亲本
| Funnel杂交
| 406 RIL
| 100K SNP
芯片,GBS | 发芽率、叶绿素
和出苗期 | GWAS,QTL
| [52]
|
| | 8亲本
| Funnel杂交
| 368 MLs
| 1000K SNP芯片,GBS | 籽粒含油量
| GWAS,QTL
| [53]
|
| | 16亲本
| Funnel杂交
| 513 RIL
| 500K SNP芯片
| 株高、叶形态、
开花期、穗大小 | GWAS
| [54]
|
| | 4亲本
| Funnel杂交
| 1044 F6
| 50K SNP芯片
| 产量、吐丝期、
株高 | QTL,AM
| [55]
|
油菜
Brassica napus | 甘蓝型
| 8亲本
| 随机杂交
| 680 F6
| –
| 始花期、抗病性
| SSD
| [56]
|
Fn表示构建F1的杂交过程和F1自交过程的总代数,Sn表示F1植株自交的代数,DH(doubled haploid)表示双单倍体系,GBS(genotyping by sequencing)表示基于测序的基因分型,QTL(quantitative trait locus)表示QTL定位,LDA(linkage disequilibrium analysis)表示连锁不平衡分析,LMC(linkage map construction)表示连锁图谱构建,AM(association mapping)表示关联作图,GWAS(genome wide association study)表示全基因组关联分析,SNP(single nucleotide polymorphisms)表示单核苷酸多态性,Funnel杂交表示“漏斗”型杂交,SSR(simple sequence repeat)表示简单重复序列,SSD(single seed descent)表示单粒传法,RIL(recombinant inbred lines)表示重组自交系,ML(lines)表示自交系,NAM(nested association map)表示巢式关联图谱 ...
... Ongom等[46]利用29个亲本的高粱MAGIC群体进行全基因组关联分析,定位到了2个已知的株高基因DWARFI和DWARF3,还检测到一个位于6号染色体上新的株高QTL.刘洪泰[47]利用8个烟草特色种质为亲本构建了一个包含600个株系并且具有丰富多样性的烟草MAGIC群体,充分发挥MAGIC群体高效、高精度和低假阳性的优势,在后代群体选育出株系株型良好、抗病且烟叶品质高的材料,作为育种中间材料或直接选育新品种. ...
Genetic properties of the MAGIC maize population:a new platform for high definition QTL mapping in Zea mays
2
2015
... Application of MAGIC group in plant genetics research
Table 1 作物
Crop | 类型
Type | 亲本选择
Parental selection | 杂交方式
Hybridization | 群体规模
Group size | 基因型鉴定
Genotyping | 研究性状
Research trait | 分析方法
Analytical method | 参考文献
Reference |
拟南芥
Arabidopsis | –
| 19亲本
| 随机杂交
| 1026 F4
| 1260 SNPs
| 抽薹期
| QTL
| [26]
|
| 水稻Rice | 籼稻 | 4亲本DC1 | 双列杂交 | 495 F6 | 6K SNP芯片 | 籽粒产量性状、穗相关性状、分蘖相关性状、抽穗期、苗期和株高
| GWAS | [23,28-31] |
| | 4亲本DC2 | 双列杂交 | 525 F6 | | | |
| | 8亲本
| Funnel杂交
| 668 F6
| 55K SNP芯片,
GBS | | |
| 籼稻 | 8亲本 | Funnel杂交 | 1328 S7 | GBS: 17387 SNP | 产量性状、稻米品质、开花期、株高、粒型、抗旱性、耐涝性、耐盐性、抗病性(稻瘟病、白叶枯病、褐斑病)
| QTL,GWAS | [32⇓-34] |
| | MAGIC PLUS | ― | 144 S8 | | | |
| | MAGIC PLUS
DH系 | ―
| 76 DH
| | | |
| 粳稻 | 8亲本 | Funnel杂交 | 500 S5 | | | |
| 籼―粳 | GLOBAL MAGIC | ― | 1402 S7 | | | |
小麦Wheat
| 春小麦
| 4亲本
| Funnel杂交
| 1579 F6
| 826 DArTs、283SNPs、53SSRs,9K SNP芯片,90K SNP芯片 | 株高、籽粒重、
胚芽鞘长度
| LMC,QTL
| [13,35-36]
|
| 冬小麦
| 8亲本
| Funnel杂交
| 1091 F7
| 90K SNP芯片
| 产量、开花期、
病害和株高 | LMC,QTL
| [37-38]
|
| 全麦粉
| 4亲本
| Funnel杂交
| 332 F6
| SSR
| 产量性状、
抗白粉病 | QTL,LMC
| [39]
|
| 欧洲
小麦 | 60亲本
| 随机杂交
| 1000 S4
| 9K SNP芯片
| 抽穗期
| LDA,GWAS
| [40]
|
| | | | | 14个KASPar SNP | | | |
大麦
Hordeum | – | 8亲本 | Funnel杂交 | 5000 DH | 9K SNP芯片 | 开花期 | QTL | [41] |
| 8亲本 | Funnel杂交 | 642 F5 | 9K SNP芯片 | 黄斑病 | LMC,QTL | [42] |
| 棉花Cotton | 陆地棉 | 11亲本 | 随机杂交 | 547 F5 | GBS | 纤维品质 | GWAS,QTL | [43] |
| | 8亲本
| 随机杂交
| 960 MLs
| SLAF-seq
| 开花期、株型、
果枝数 | GWAS,QTL
| [44]
|
| | 11亲本 | 随机杂交 | 550 RIL | 47K SNP芯片 | 株高、茎秆质量 | GWAS,QTL | [45] |
高粱
Sorghum | –
| 29亲本
| 随机杂交
| 1000 F6
| GBS
| 株高
| GWAS,QTL
| [46]
|
作物
Crop | 类型
Type | 亲本选择
Parental selection | 杂交方式
Hybridization | 群体规模
Group size | 基因型鉴定
Genotyping | 研究性状
Research trait | 分析方法
Analytical method | 参考文献
Reference |
烟草
Tobacco | –
| 8亲本
| 双列杂交
| 600 F4
| 430K 芯片
| 株型、烟叶品质
| GWAS
| [47]
|
玉米
Maize | –
| 8亲本
| Funnel杂交
| 1636 F6
| GBS
| 开花期、株高、
穗位高 | LMC,AM,QTL | [48]
|
| | 4亲本
| Funnel杂交
| 1149 S3
| 118K SNP 芯片
| 株高、穗高、散
粉期、吐丝期 | QTL,GWAS
| [49]
|
| | 8亲本
| Funnel杂交
| 401 RIL
| 56K SNP 芯片
| 苗重、苗长和
黄萎病 | QTL,NAM
| [50]
|
| | 8亲本
| Funnel杂交
| 700F6
| 110K SNP
芯片 | 镰孢菌穗腐病
| LMC,AM,QTL | [51]
|
| | 8亲本
| Funnel杂交
| 406 RIL
| 100K SNP
芯片,GBS | 发芽率、叶绿素
和出苗期 | GWAS,QTL
| [52]
|
| | 8亲本
| Funnel杂交
| 368 MLs
| 1000K SNP芯片,GBS | 籽粒含油量
| GWAS,QTL
| [53]
|
| | 16亲本
| Funnel杂交
| 513 RIL
| 500K SNP芯片
| 株高、叶形态、
开花期、穗大小 | GWAS
| [54]
|
| | 4亲本
| Funnel杂交
| 1044 F6
| 50K SNP芯片
| 产量、吐丝期、
株高 | QTL,AM
| [55]
|
油菜
Brassica napus | 甘蓝型
| 8亲本
| 随机杂交
| 680 F6
| –
| 始花期、抗病性
| SSD
| [56]
|
Fn表示构建F1的杂交过程和F1自交过程的总代数,Sn表示F1植株自交的代数,DH(doubled haploid)表示双单倍体系,GBS(genotyping by sequencing)表示基于测序的基因分型,QTL(quantitative trait locus)表示QTL定位,LDA(linkage disequilibrium analysis)表示连锁不平衡分析,LMC(linkage map construction)表示连锁图谱构建,AM(association mapping)表示关联作图,GWAS(genome wide association study)表示全基因组关联分析,SNP(single nucleotide polymorphisms)表示单核苷酸多态性,Funnel杂交表示“漏斗”型杂交,SSR(simple sequence repeat)表示简单重复序列,SSD(single seed descent)表示单粒传法,RIL(recombinant inbred lines)表示重组自交系,ML(lines)表示自交系,NAM(nested association map)表示巢式关联图谱 ...
... 在玉米MAGIC群体遗传研究中,Dell'Acqua等[48]在玉米中构建了一个含1636个家系的8亲本MAGIC群体(图2).在群体构建过程中包含B96×HP301 F1的四元杂交由于花期不遇而构建失败,所以作者引入第9个亲本(CML91)进行B73×CML91双向杂交以弥补失败的B96×HP301的四元杂交.此研究使用芯片对529个MAGIC家系鉴定基因型得到54 234个SNPs标记.使用连锁分析和关联分析对开花期、株高、穗位高和籽粒产量4个表型进行了QTL扫描.同时考虑亲本的基因组数据和转录组数据分别预测了3个开花期和3个籽粒产量的候选基因. ...
Four parent maize (FPM) population:effects of mating designs on linkage disequillibrium and mapping quantitative traits
2
2018
... Application of MAGIC group in plant genetics research
Table 1 作物
Crop | 类型
Type | 亲本选择
Parental selection | 杂交方式
Hybridization | 群体规模
Group size | 基因型鉴定
Genotyping | 研究性状
Research trait | 分析方法
Analytical method | 参考文献
Reference |
拟南芥
Arabidopsis | –
| 19亲本
| 随机杂交
| 1026 F4
| 1260 SNPs
| 抽薹期
| QTL
| [26]
|
| 水稻Rice | 籼稻 | 4亲本DC1 | 双列杂交 | 495 F6 | 6K SNP芯片 | 籽粒产量性状、穗相关性状、分蘖相关性状、抽穗期、苗期和株高
| GWAS | [23,28-31] |
| | 4亲本DC2 | 双列杂交 | 525 F6 | | | |
| | 8亲本
| Funnel杂交
| 668 F6
| 55K SNP芯片,
GBS | | |
| 籼稻 | 8亲本 | Funnel杂交 | 1328 S7 | GBS: 17387 SNP | 产量性状、稻米品质、开花期、株高、粒型、抗旱性、耐涝性、耐盐性、抗病性(稻瘟病、白叶枯病、褐斑病)
| QTL,GWAS | [32⇓-34] |
| | MAGIC PLUS | ― | 144 S8 | | | |
| | MAGIC PLUS
DH系 | ―
| 76 DH
| | | |
| 粳稻 | 8亲本 | Funnel杂交 | 500 S5 | | | |
| 籼―粳 | GLOBAL MAGIC | ― | 1402 S7 | | | |
小麦Wheat
| 春小麦
| 4亲本
| Funnel杂交
| 1579 F6
| 826 DArTs、283SNPs、53SSRs,9K SNP芯片,90K SNP芯片 | 株高、籽粒重、
胚芽鞘长度
| LMC,QTL
| [13,35-36]
|
| 冬小麦
| 8亲本
| Funnel杂交
| 1091 F7
| 90K SNP芯片
| 产量、开花期、
病害和株高 | LMC,QTL
| [37-38]
|
| 全麦粉
| 4亲本
| Funnel杂交
| 332 F6
| SSR
| 产量性状、
抗白粉病 | QTL,LMC
| [39]
|
| 欧洲
小麦 | 60亲本
| 随机杂交
| 1000 S4
| 9K SNP芯片
| 抽穗期
| LDA,GWAS
| [40]
|
| | | | | 14个KASPar SNP | | | |
大麦
Hordeum | – | 8亲本 | Funnel杂交 | 5000 DH | 9K SNP芯片 | 开花期 | QTL | [41] |
| 8亲本 | Funnel杂交 | 642 F5 | 9K SNP芯片 | 黄斑病 | LMC,QTL | [42] |
| 棉花Cotton | 陆地棉 | 11亲本 | 随机杂交 | 547 F5 | GBS | 纤维品质 | GWAS,QTL | [43] |
| | 8亲本
| 随机杂交
| 960 MLs
| SLAF-seq
| 开花期、株型、
果枝数 | GWAS,QTL
| [44]
|
| | 11亲本 | 随机杂交 | 550 RIL | 47K SNP芯片 | 株高、茎秆质量 | GWAS,QTL | [45] |
高粱
Sorghum | –
| 29亲本
| 随机杂交
| 1000 F6
| GBS
| 株高
| GWAS,QTL
| [46]
|
作物
Crop | 类型
Type | 亲本选择
Parental selection | 杂交方式
Hybridization | 群体规模
Group size | 基因型鉴定
Genotyping | 研究性状
Research trait | 分析方法
Analytical method | 参考文献
Reference |
烟草
Tobacco | –
| 8亲本
| 双列杂交
| 600 F4
| 430K 芯片
| 株型、烟叶品质
| GWAS
| [47]
|
玉米
Maize | –
| 8亲本
| Funnel杂交
| 1636 F6
| GBS
| 开花期、株高、
穗位高 | LMC,AM,QTL | [48]
|
| | 4亲本
| Funnel杂交
| 1149 S3
| 118K SNP 芯片
| 株高、穗高、散
粉期、吐丝期 | QTL,GWAS
| [49]
|
| | 8亲本
| Funnel杂交
| 401 RIL
| 56K SNP 芯片
| 苗重、苗长和
黄萎病 | QTL,NAM
| [50]
|
| | 8亲本
| Funnel杂交
| 700F6
| 110K SNP
芯片 | 镰孢菌穗腐病
| LMC,AM,QTL | [51]
|
| | 8亲本
| Funnel杂交
| 406 RIL
| 100K SNP
芯片,GBS | 发芽率、叶绿素
和出苗期 | GWAS,QTL
| [52]
|
| | 8亲本
| Funnel杂交
| 368 MLs
| 1000K SNP芯片,GBS | 籽粒含油量
| GWAS,QTL
| [53]
|
| | 16亲本
| Funnel杂交
| 513 RIL
| 500K SNP芯片
| 株高、叶形态、
开花期、穗大小 | GWAS
| [54]
|
| | 4亲本
| Funnel杂交
| 1044 F6
| 50K SNP芯片
| 产量、吐丝期、
株高 | QTL,AM
| [55]
|
油菜
Brassica napus | 甘蓝型
| 8亲本
| 随机杂交
| 680 F6
| –
| 始花期、抗病性
| SSD
| [56]
|
Fn表示构建F1的杂交过程和F1自交过程的总代数,Sn表示F1植株自交的代数,DH(doubled haploid)表示双单倍体系,GBS(genotyping by sequencing)表示基于测序的基因分型,QTL(quantitative trait locus)表示QTL定位,LDA(linkage disequilibrium analysis)表示连锁不平衡分析,LMC(linkage map construction)表示连锁图谱构建,AM(association mapping)表示关联作图,GWAS(genome wide association study)表示全基因组关联分析,SNP(single nucleotide polymorphisms)表示单核苷酸多态性,Funnel杂交表示“漏斗”型杂交,SSR(simple sequence repeat)表示简单重复序列,SSD(single seed descent)表示单粒传法,RIL(recombinant inbred lines)表示重组自交系,ML(lines)表示自交系,NAM(nested association map)表示巢式关联图谱 ...
... Anderson等[49]通过增加等位基因的多样性和有效的基因重组,发现MAGIC群体可以提供更好的遗传定位分辨率.作者采用4亲本玉米MAGIC群体和双亲本群体进行5种不同的交配设计,比较了QTL检测能力和遗传分辨率.该群体共有1149个株系,有118 509个遗传标记.并在7个环境中对株高(PH)、穗位高(EH)、散粉期(DTA)和吐丝期(DTS)进行了连续3年的测定.连锁不平衡(LD)分析表明,与双亲群体相比,4个亲本杂交第3代(4way3sib)的遗传分辨率提高了3~4倍(r2 < 0.80),LD衰退程度降低了2.5倍(r2 < 0.20).群体功能模拟表明,对QTL检测能力的影响更大的是样本量,而不是交配设计.利用关联映射软件分别鉴定出2、5、7和6个PH、EH、DTA和DTS性状的QTL. ...
Unravelling the genetic basis of Fusariom seedling rot resistance in the MAGIC maize population:novel targets for breeding
2
2019
... Application of MAGIC group in plant genetics research
Table 1 作物
Crop | 类型
Type | 亲本选择
Parental selection | 杂交方式
Hybridization | 群体规模
Group size | 基因型鉴定
Genotyping | 研究性状
Research trait | 分析方法
Analytical method | 参考文献
Reference |
拟南芥
Arabidopsis | –
| 19亲本
| 随机杂交
| 1026 F4
| 1260 SNPs
| 抽薹期
| QTL
| [26]
|
| 水稻Rice | 籼稻 | 4亲本DC1 | 双列杂交 | 495 F6 | 6K SNP芯片 | 籽粒产量性状、穗相关性状、分蘖相关性状、抽穗期、苗期和株高
| GWAS | [23,28-31] |
| | 4亲本DC2 | 双列杂交 | 525 F6 | | | |
| | 8亲本
| Funnel杂交
| 668 F6
| 55K SNP芯片,
GBS | | |
| 籼稻 | 8亲本 | Funnel杂交 | 1328 S7 | GBS: 17387 SNP | 产量性状、稻米品质、开花期、株高、粒型、抗旱性、耐涝性、耐盐性、抗病性(稻瘟病、白叶枯病、褐斑病)
| QTL,GWAS | [32⇓-34] |
| | MAGIC PLUS | ― | 144 S8 | | | |
| | MAGIC PLUS
DH系 | ―
| 76 DH
| | | |
| 粳稻 | 8亲本 | Funnel杂交 | 500 S5 | | | |
| 籼―粳 | GLOBAL MAGIC | ― | 1402 S7 | | | |
小麦Wheat
| 春小麦
| 4亲本
| Funnel杂交
| 1579 F6
| 826 DArTs、283SNPs、53SSRs,9K SNP芯片,90K SNP芯片 | 株高、籽粒重、
胚芽鞘长度
| LMC,QTL
| [13,35-36]
|
| 冬小麦
| 8亲本
| Funnel杂交
| 1091 F7
| 90K SNP芯片
| 产量、开花期、
病害和株高 | LMC,QTL
| [37-38]
|
| 全麦粉
| 4亲本
| Funnel杂交
| 332 F6
| SSR
| 产量性状、
抗白粉病 | QTL,LMC
| [39]
|
| 欧洲
小麦 | 60亲本
| 随机杂交
| 1000 S4
| 9K SNP芯片
| 抽穗期
| LDA,GWAS
| [40]
|
| | | | | 14个KASPar SNP | | | |
大麦
Hordeum | – | 8亲本 | Funnel杂交 | 5000 DH | 9K SNP芯片 | 开花期 | QTL | [41] |
| 8亲本 | Funnel杂交 | 642 F5 | 9K SNP芯片 | 黄斑病 | LMC,QTL | [42] |
| 棉花Cotton | 陆地棉 | 11亲本 | 随机杂交 | 547 F5 | GBS | 纤维品质 | GWAS,QTL | [43] |
| | 8亲本
| 随机杂交
| 960 MLs
| SLAF-seq
| 开花期、株型、
果枝数 | GWAS,QTL
| [44]
|
| | 11亲本 | 随机杂交 | 550 RIL | 47K SNP芯片 | 株高、茎秆质量 | GWAS,QTL | [45] |
高粱
Sorghum | –
| 29亲本
| 随机杂交
| 1000 F6
| GBS
| 株高
| GWAS,QTL
| [46]
|
作物
Crop | 类型
Type | 亲本选择
Parental selection | 杂交方式
Hybridization | 群体规模
Group size | 基因型鉴定
Genotyping | 研究性状
Research trait | 分析方法
Analytical method | 参考文献
Reference |
烟草
Tobacco | –
| 8亲本
| 双列杂交
| 600 F4
| 430K 芯片
| 株型、烟叶品质
| GWAS
| [47]
|
玉米
Maize | –
| 8亲本
| Funnel杂交
| 1636 F6
| GBS
| 开花期、株高、
穗位高 | LMC,AM,QTL | [48]
|
| | 4亲本
| Funnel杂交
| 1149 S3
| 118K SNP 芯片
| 株高、穗高、散
粉期、吐丝期 | QTL,GWAS
| [49]
|
| | 8亲本
| Funnel杂交
| 401 RIL
| 56K SNP 芯片
| 苗重、苗长和
黄萎病 | QTL,NAM
| [50]
|
| | 8亲本
| Funnel杂交
| 700F6
| 110K SNP
芯片 | 镰孢菌穗腐病
| LMC,AM,QTL | [51]
|
| | 8亲本
| Funnel杂交
| 406 RIL
| 100K SNP
芯片,GBS | 发芽率、叶绿素
和出苗期 | GWAS,QTL
| [52]
|
| | 8亲本
| Funnel杂交
| 368 MLs
| 1000K SNP芯片,GBS | 籽粒含油量
| GWAS,QTL
| [53]
|
| | 16亲本
| Funnel杂交
| 513 RIL
| 500K SNP芯片
| 株高、叶形态、
开花期、穗大小 | GWAS
| [54]
|
| | 4亲本
| Funnel杂交
| 1044 F6
| 50K SNP芯片
| 产量、吐丝期、
株高 | QTL,AM
| [55]
|
油菜
Brassica napus | 甘蓝型
| 8亲本
| 随机杂交
| 680 F6
| –
| 始花期、抗病性
| SSD
| [56]
|
Fn表示构建F1的杂交过程和F1自交过程的总代数,Sn表示F1植株自交的代数,DH(doubled haploid)表示双单倍体系,GBS(genotyping by sequencing)表示基于测序的基因分型,QTL(quantitative trait locus)表示QTL定位,LDA(linkage disequilibrium analysis)表示连锁不平衡分析,LMC(linkage map construction)表示连锁图谱构建,AM(association mapping)表示关联作图,GWAS(genome wide association study)表示全基因组关联分析,SNP(single nucleotide polymorphisms)表示单核苷酸多态性,Funnel杂交表示“漏斗”型杂交,SSR(simple sequence repeat)表示简单重复序列,SSD(single seed descent)表示单粒传法,RIL(recombinant inbred lines)表示重组自交系,ML(lines)表示自交系,NAM(nested association map)表示巢式关联图谱 ...
... Septiani等[50]以玉米为研究对象,利用26个自交系的多样性,先建立了一个高度多样性的NAM群体,然后选择8个不同的玉米自交系构建MAGIC群体.该研究利用Illumina 56 110个SNP标记对MAGIC群体进行基因分型.利用MAGIC群体,用卷巾法(rolled towel assay,RTA)对镰刀菌腐苗(fusarium seedling rot,FSR)抗性进行了高分辨率的QTL定位,测定了401份MAGIC玉米重组自交系的侵染严重程度、苗重和苗长,并对RIL单倍型进行QTL定位,鉴定出10个QTL.对鉴定出的FSR QTL中8个候选基因进行了鉴定,本研究将RTA法用于玉米MAGIC群体,对鉴定玉米黄萎病早期抗性QTL和候选基因提供了一种经济而又快速的方法. ...
QTLs for resistance to fusarium ear rot in a multiparent advanced generation intercross (MAGIC) maize population
2
2019
... Application of MAGIC group in plant genetics research
Table 1 作物
Crop | 类型
Type | 亲本选择
Parental selection | 杂交方式
Hybridization | 群体规模
Group size | 基因型鉴定
Genotyping | 研究性状
Research trait | 分析方法
Analytical method | 参考文献
Reference |
拟南芥
Arabidopsis | –
| 19亲本
| 随机杂交
| 1026 F4
| 1260 SNPs
| 抽薹期
| QTL
| [26]
|
| 水稻Rice | 籼稻 | 4亲本DC1 | 双列杂交 | 495 F6 | 6K SNP芯片 | 籽粒产量性状、穗相关性状、分蘖相关性状、抽穗期、苗期和株高
| GWAS | [23,28-31] |
| | 4亲本DC2 | 双列杂交 | 525 F6 | | | |
| | 8亲本
| Funnel杂交
| 668 F6
| 55K SNP芯片,
GBS | | |
| 籼稻 | 8亲本 | Funnel杂交 | 1328 S7 | GBS: 17387 SNP | 产量性状、稻米品质、开花期、株高、粒型、抗旱性、耐涝性、耐盐性、抗病性(稻瘟病、白叶枯病、褐斑病)
| QTL,GWAS | [32⇓-34] |
| | MAGIC PLUS | ― | 144 S8 | | | |
| | MAGIC PLUS
DH系 | ―
| 76 DH
| | | |
| 粳稻 | 8亲本 | Funnel杂交 | 500 S5 | | | |
| 籼―粳 | GLOBAL MAGIC | ― | 1402 S7 | | | |
小麦Wheat
| 春小麦
| 4亲本
| Funnel杂交
| 1579 F6
| 826 DArTs、283SNPs、53SSRs,9K SNP芯片,90K SNP芯片 | 株高、籽粒重、
胚芽鞘长度
| LMC,QTL
| [13,35-36]
|
| 冬小麦
| 8亲本
| Funnel杂交
| 1091 F7
| 90K SNP芯片
| 产量、开花期、
病害和株高 | LMC,QTL
| [37-38]
|
| 全麦粉
| 4亲本
| Funnel杂交
| 332 F6
| SSR
| 产量性状、
抗白粉病 | QTL,LMC
| [39]
|
| 欧洲
小麦 | 60亲本
| 随机杂交
| 1000 S4
| 9K SNP芯片
| 抽穗期
| LDA,GWAS
| [40]
|
| | | | | 14个KASPar SNP | | | |
大麦
Hordeum | – | 8亲本 | Funnel杂交 | 5000 DH | 9K SNP芯片 | 开花期 | QTL | [41] |
| 8亲本 | Funnel杂交 | 642 F5 | 9K SNP芯片 | 黄斑病 | LMC,QTL | [42] |
| 棉花Cotton | 陆地棉 | 11亲本 | 随机杂交 | 547 F5 | GBS | 纤维品质 | GWAS,QTL | [43] |
| | 8亲本
| 随机杂交
| 960 MLs
| SLAF-seq
| 开花期、株型、
果枝数 | GWAS,QTL
| [44]
|
| | 11亲本 | 随机杂交 | 550 RIL | 47K SNP芯片 | 株高、茎秆质量 | GWAS,QTL | [45] |
高粱
Sorghum | –
| 29亲本
| 随机杂交
| 1000 F6
| GBS
| 株高
| GWAS,QTL
| [46]
|
作物
Crop | 类型
Type | 亲本选择
Parental selection | 杂交方式
Hybridization | 群体规模
Group size | 基因型鉴定
Genotyping | 研究性状
Research trait | 分析方法
Analytical method | 参考文献
Reference |
烟草
Tobacco | –
| 8亲本
| 双列杂交
| 600 F4
| 430K 芯片
| 株型、烟叶品质
| GWAS
| [47]
|
玉米
Maize | –
| 8亲本
| Funnel杂交
| 1636 F6
| GBS
| 开花期、株高、
穗位高 | LMC,AM,QTL | [48]
|
| | 4亲本
| Funnel杂交
| 1149 S3
| 118K SNP 芯片
| 株高、穗高、散
粉期、吐丝期 | QTL,GWAS
| [49]
|
| | 8亲本
| Funnel杂交
| 401 RIL
| 56K SNP 芯片
| 苗重、苗长和
黄萎病 | QTL,NAM
| [50]
|
| | 8亲本
| Funnel杂交
| 700F6
| 110K SNP
芯片 | 镰孢菌穗腐病
| LMC,AM,QTL | [51]
|
| | 8亲本
| Funnel杂交
| 406 RIL
| 100K SNP
芯片,GBS | 发芽率、叶绿素
和出苗期 | GWAS,QTL
| [52]
|
| | 8亲本
| Funnel杂交
| 368 MLs
| 1000K SNP芯片,GBS | 籽粒含油量
| GWAS,QTL
| [53]
|
| | 16亲本
| Funnel杂交
| 513 RIL
| 500K SNP芯片
| 株高、叶形态、
开花期、穗大小 | GWAS
| [54]
|
| | 4亲本
| Funnel杂交
| 1044 F6
| 50K SNP芯片
| 产量、吐丝期、
株高 | QTL,AM
| [55]
|
油菜
Brassica napus | 甘蓝型
| 8亲本
| 随机杂交
| 680 F6
| –
| 始花期、抗病性
| SSD
| [56]
|
Fn表示构建F1的杂交过程和F1自交过程的总代数,Sn表示F1植株自交的代数,DH(doubled haploid)表示双单倍体系,GBS(genotyping by sequencing)表示基于测序的基因分型,QTL(quantitative trait locus)表示QTL定位,LDA(linkage disequilibrium analysis)表示连锁不平衡分析,LMC(linkage map construction)表示连锁图谱构建,AM(association mapping)表示关联作图,GWAS(genome wide association study)表示全基因组关联分析,SNP(single nucleotide polymorphisms)表示单核苷酸多态性,Funnel杂交表示“漏斗”型杂交,SSR(simple sequence repeat)表示简单重复序列,SSD(single seed descent)表示单粒传法,RIL(recombinant inbred lines)表示重组自交系,ML(lines)表示自交系,NAM(nested association map)表示巢式关联图谱 ...
... Butrón等[51]利用玉米近交系进行连锁和关联作图的替代方法深入研究与抗镰孢菌穗腐病相关的QTL.通过对玉米MAGIC群体检测与镰孢菌穗腐病抗性相关的QTL适用性的研究,表明MAGIC群体可以作为不同近交系群体的有效补充,并且发现在温带材料中含有抗性相关基因座的基因组区域.同时发现镰孢菌穗腐病抗性的有利等位基因频率一般都较高,这些发现证实了对镰孢菌穗腐病抗性的数量特征,其中许多基因座具有加性效应.证明在关键区域如3号染色体的210~220Mb和7号染色体的166~173Mb含有镰孢菌穗腐病抗性QTL. ...
Dissecting the genetics of cold tolerance in a multiparental maize population
2
2020
... Application of MAGIC group in plant genetics research
Table 1 作物
Crop | 类型
Type | 亲本选择
Parental selection | 杂交方式
Hybridization | 群体规模
Group size | 基因型鉴定
Genotyping | 研究性状
Research trait | 分析方法
Analytical method | 参考文献
Reference |
拟南芥
Arabidopsis | –
| 19亲本
| 随机杂交
| 1026 F4
| 1260 SNPs
| 抽薹期
| QTL
| [26]
|
| 水稻Rice | 籼稻 | 4亲本DC1 | 双列杂交 | 495 F6 | 6K SNP芯片 | 籽粒产量性状、穗相关性状、分蘖相关性状、抽穗期、苗期和株高
| GWAS | [23,28-31] |
| | 4亲本DC2 | 双列杂交 | 525 F6 | | | |
| | 8亲本
| Funnel杂交
| 668 F6
| 55K SNP芯片,
GBS | | |
| 籼稻 | 8亲本 | Funnel杂交 | 1328 S7 | GBS: 17387 SNP | 产量性状、稻米品质、开花期、株高、粒型、抗旱性、耐涝性、耐盐性、抗病性(稻瘟病、白叶枯病、褐斑病)
| QTL,GWAS | [32⇓-34] |
| | MAGIC PLUS | ― | 144 S8 | | | |
| | MAGIC PLUS
DH系 | ―
| 76 DH
| | | |
| 粳稻 | 8亲本 | Funnel杂交 | 500 S5 | | | |
| 籼―粳 | GLOBAL MAGIC | ― | 1402 S7 | | | |
小麦Wheat
| 春小麦
| 4亲本
| Funnel杂交
| 1579 F6
| 826 DArTs、283SNPs、53SSRs,9K SNP芯片,90K SNP芯片 | 株高、籽粒重、
胚芽鞘长度
| LMC,QTL
| [13,35-36]
|
| 冬小麦
| 8亲本
| Funnel杂交
| 1091 F7
| 90K SNP芯片
| 产量、开花期、
病害和株高 | LMC,QTL
| [37-38]
|
| 全麦粉
| 4亲本
| Funnel杂交
| 332 F6
| SSR
| 产量性状、
抗白粉病 | QTL,LMC
| [39]
|
| 欧洲
小麦 | 60亲本
| 随机杂交
| 1000 S4
| 9K SNP芯片
| 抽穗期
| LDA,GWAS
| [40]
|
| | | | | 14个KASPar SNP | | | |
大麦
Hordeum | – | 8亲本 | Funnel杂交 | 5000 DH | 9K SNP芯片 | 开花期 | QTL | [41] |
| 8亲本 | Funnel杂交 | 642 F5 | 9K SNP芯片 | 黄斑病 | LMC,QTL | [42] |
| 棉花Cotton | 陆地棉 | 11亲本 | 随机杂交 | 547 F5 | GBS | 纤维品质 | GWAS,QTL | [43] |
| | 8亲本
| 随机杂交
| 960 MLs
| SLAF-seq
| 开花期、株型、
果枝数 | GWAS,QTL
| [44]
|
| | 11亲本 | 随机杂交 | 550 RIL | 47K SNP芯片 | 株高、茎秆质量 | GWAS,QTL | [45] |
高粱
Sorghum | –
| 29亲本
| 随机杂交
| 1000 F6
| GBS
| 株高
| GWAS,QTL
| [46]
|
作物
Crop | 类型
Type | 亲本选择
Parental selection | 杂交方式
Hybridization | 群体规模
Group size | 基因型鉴定
Genotyping | 研究性状
Research trait | 分析方法
Analytical method | 参考文献
Reference |
烟草
Tobacco | –
| 8亲本
| 双列杂交
| 600 F4
| 430K 芯片
| 株型、烟叶品质
| GWAS
| [47]
|
玉米
Maize | –
| 8亲本
| Funnel杂交
| 1636 F6
| GBS
| 开花期、株高、
穗位高 | LMC,AM,QTL | [48]
|
| | 4亲本
| Funnel杂交
| 1149 S3
| 118K SNP 芯片
| 株高、穗高、散
粉期、吐丝期 | QTL,GWAS
| [49]
|
| | 8亲本
| Funnel杂交
| 401 RIL
| 56K SNP 芯片
| 苗重、苗长和
黄萎病 | QTL,NAM
| [50]
|
| | 8亲本
| Funnel杂交
| 700F6
| 110K SNP
芯片 | 镰孢菌穗腐病
| LMC,AM,QTL | [51]
|
| | 8亲本
| Funnel杂交
| 406 RIL
| 100K SNP
芯片,GBS | 发芽率、叶绿素
和出苗期 | GWAS,QTL
| [52]
|
| | 8亲本
| Funnel杂交
| 368 MLs
| 1000K SNP芯片,GBS | 籽粒含油量
| GWAS,QTL
| [53]
|
| | 16亲本
| Funnel杂交
| 513 RIL
| 500K SNP芯片
| 株高、叶形态、
开花期、穗大小 | GWAS
| [54]
|
| | 4亲本
| Funnel杂交
| 1044 F6
| 50K SNP芯片
| 产量、吐丝期、
株高 | QTL,AM
| [55]
|
油菜
Brassica napus | 甘蓝型
| 8亲本
| 随机杂交
| 680 F6
| –
| 始花期、抗病性
| SSD
| [56]
|
Fn表示构建F1的杂交过程和F1自交过程的总代数,Sn表示F1植株自交的代数,DH(doubled haploid)表示双单倍体系,GBS(genotyping by sequencing)表示基于测序的基因分型,QTL(quantitative trait locus)表示QTL定位,LDA(linkage disequilibrium analysis)表示连锁不平衡分析,LMC(linkage map construction)表示连锁图谱构建,AM(association mapping)表示关联作图,GWAS(genome wide association study)表示全基因组关联分析,SNP(single nucleotide polymorphisms)表示单核苷酸多态性,Funnel杂交表示“漏斗”型杂交,SSR(simple sequence repeat)表示简单重复序列,SSD(single seed descent)表示单粒传法,RIL(recombinant inbred lines)表示重组自交系,ML(lines)表示自交系,NAM(nested association map)表示巢式关联图谱 ...
... 环境对耐逆性的鉴定和遗传定位会有很大的影响,因此,在MAGIC群体构建的基础上,进行多年多点的耐逆鉴定非常有必要,同时可以分析环境与基因型之间的互作.Yi等[52]以8亲本的406个MAGIC群体的重组自交系为材料,分别在温室低温、田间早播和正常播种条件下进行耐低温胁迫的鉴定,记录群体播种至出苗天数、叶绿素含量和最大光化学量子产量(Fv/Fm).与温室和田间寒冷相比,正常播种条件下的发芽率、叶绿素含量、Fv/Fm和干重较高,出苗时间推迟.在所有环境中,MAGIC群体的亲本和RIL之间的幼苗性状均存在显著相关,且大多数是在冷害和早播条件下发现的.关联分析结果显示,858个SNP与所有性状均存在显著相关,且大多数是在冷害和早播条件下发现.这些结果表明,MAGIC群体是进行多环境下耐冷性研究的有效工具. ...
Genome-wide association study dissects the genetic architecture of oil biosynthesis in maize kernels
1
2013
... Application of MAGIC group in plant genetics research
Table 1 作物
Crop | 类型
Type | 亲本选择
Parental selection | 杂交方式
Hybridization | 群体规模
Group size | 基因型鉴定
Genotyping | 研究性状
Research trait | 分析方法
Analytical method | 参考文献
Reference |
拟南芥
Arabidopsis | –
| 19亲本
| 随机杂交
| 1026 F4
| 1260 SNPs
| 抽薹期
| QTL
| [26]
|
| 水稻Rice | 籼稻 | 4亲本DC1 | 双列杂交 | 495 F6 | 6K SNP芯片 | 籽粒产量性状、穗相关性状、分蘖相关性状、抽穗期、苗期和株高
| GWAS | [23,28-31] |
| | 4亲本DC2 | 双列杂交 | 525 F6 | | | |
| | 8亲本
| Funnel杂交
| 668 F6
| 55K SNP芯片,
GBS | | |
| 籼稻 | 8亲本 | Funnel杂交 | 1328 S7 | GBS: 17387 SNP | 产量性状、稻米品质、开花期、株高、粒型、抗旱性、耐涝性、耐盐性、抗病性(稻瘟病、白叶枯病、褐斑病)
| QTL,GWAS | [32⇓-34] |
| | MAGIC PLUS | ― | 144 S8 | | | |
| | MAGIC PLUS
DH系 | ―
| 76 DH
| | | |
| 粳稻 | 8亲本 | Funnel杂交 | 500 S5 | | | |
| 籼―粳 | GLOBAL MAGIC | ― | 1402 S7 | | | |
小麦Wheat
| 春小麦
| 4亲本
| Funnel杂交
| 1579 F6
| 826 DArTs、283SNPs、53SSRs,9K SNP芯片,90K SNP芯片 | 株高、籽粒重、
胚芽鞘长度
| LMC,QTL
| [13,35-36]
|
| 冬小麦
| 8亲本
| Funnel杂交
| 1091 F7
| 90K SNP芯片
| 产量、开花期、
病害和株高 | LMC,QTL
| [37-38]
|
| 全麦粉
| 4亲本
| Funnel杂交
| 332 F6
| SSR
| 产量性状、
抗白粉病 | QTL,LMC
| [39]
|
| 欧洲
小麦 | 60亲本
| 随机杂交
| 1000 S4
| 9K SNP芯片
| 抽穗期
| LDA,GWAS
| [40]
|
| | | | | 14个KASPar SNP | | | |
大麦
Hordeum | – | 8亲本 | Funnel杂交 | 5000 DH | 9K SNP芯片 | 开花期 | QTL | [41] |
| 8亲本 | Funnel杂交 | 642 F5 | 9K SNP芯片 | 黄斑病 | LMC,QTL | [42] |
| 棉花Cotton | 陆地棉 | 11亲本 | 随机杂交 | 547 F5 | GBS | 纤维品质 | GWAS,QTL | [43] |
| | 8亲本
| 随机杂交
| 960 MLs
| SLAF-seq
| 开花期、株型、
果枝数 | GWAS,QTL
| [44]
|
| | 11亲本 | 随机杂交 | 550 RIL | 47K SNP芯片 | 株高、茎秆质量 | GWAS,QTL | [45] |
高粱
Sorghum | –
| 29亲本
| 随机杂交
| 1000 F6
| GBS
| 株高
| GWAS,QTL
| [46]
|
作物
Crop | 类型
Type | 亲本选择
Parental selection | 杂交方式
Hybridization | 群体规模
Group size | 基因型鉴定
Genotyping | 研究性状
Research trait | 分析方法
Analytical method | 参考文献
Reference |
烟草
Tobacco | –
| 8亲本
| 双列杂交
| 600 F4
| 430K 芯片
| 株型、烟叶品质
| GWAS
| [47]
|
玉米
Maize | –
| 8亲本
| Funnel杂交
| 1636 F6
| GBS
| 开花期、株高、
穗位高 | LMC,AM,QTL | [48]
|
| | 4亲本
| Funnel杂交
| 1149 S3
| 118K SNP 芯片
| 株高、穗高、散
粉期、吐丝期 | QTL,GWAS
| [49]
|
| | 8亲本
| Funnel杂交
| 401 RIL
| 56K SNP 芯片
| 苗重、苗长和
黄萎病 | QTL,NAM
| [50]
|
| | 8亲本
| Funnel杂交
| 700F6
| 110K SNP
芯片 | 镰孢菌穗腐病
| LMC,AM,QTL | [51]
|
| | 8亲本
| Funnel杂交
| 406 RIL
| 100K SNP
芯片,GBS | 发芽率、叶绿素
和出苗期 | GWAS,QTL
| [52]
|
| | 8亲本
| Funnel杂交
| 368 MLs
| 1000K SNP芯片,GBS | 籽粒含油量
| GWAS,QTL
| [53]
|
| | 16亲本
| Funnel杂交
| 513 RIL
| 500K SNP芯片
| 株高、叶形态、
开花期、穗大小 | GWAS
| [54]
|
| | 4亲本
| Funnel杂交
| 1044 F6
| 50K SNP芯片
| 产量、吐丝期、
株高 | QTL,AM
| [55]
|
油菜
Brassica napus | 甘蓝型
| 8亲本
| 随机杂交
| 680 F6
| –
| 始花期、抗病性
| SSD
| [56]
|
Fn表示构建F1的杂交过程和F1自交过程的总代数,Sn表示F1植株自交的代数,DH(doubled haploid)表示双单倍体系,GBS(genotyping by sequencing)表示基于测序的基因分型,QTL(quantitative trait locus)表示QTL定位,LDA(linkage disequilibrium analysis)表示连锁不平衡分析,LMC(linkage map construction)表示连锁图谱构建,AM(association mapping)表示关联作图,GWAS(genome wide association study)表示全基因组关联分析,SNP(single nucleotide polymorphisms)表示单核苷酸多态性,Funnel杂交表示“漏斗”型杂交,SSR(simple sequence repeat)表示简单重复序列,SSD(single seed descent)表示单粒传法,RIL(recombinant inbred lines)表示重组自交系,ML(lines)表示自交系,NAM(nested association map)表示巢式关联图谱 ...
Genome wide association studies using a new nonparametric model reveal the genetic architecture of 17 agronomic traits in an enlarged maize association panel
1
2014
... Application of MAGIC group in plant genetics research
Table 1 作物
Crop | 类型
Type | 亲本选择
Parental selection | 杂交方式
Hybridization | 群体规模
Group size | 基因型鉴定
Genotyping | 研究性状
Research trait | 分析方法
Analytical method | 参考文献
Reference |
拟南芥
Arabidopsis | –
| 19亲本
| 随机杂交
| 1026 F4
| 1260 SNPs
| 抽薹期
| QTL
| [26]
|
| 水稻Rice | 籼稻 | 4亲本DC1 | 双列杂交 | 495 F6 | 6K SNP芯片 | 籽粒产量性状、穗相关性状、分蘖相关性状、抽穗期、苗期和株高
| GWAS | [23,28-31] |
| | 4亲本DC2 | 双列杂交 | 525 F6 | | | |
| | 8亲本
| Funnel杂交
| 668 F6
| 55K SNP芯片,
GBS | | |
| 籼稻 | 8亲本 | Funnel杂交 | 1328 S7 | GBS: 17387 SNP | 产量性状、稻米品质、开花期、株高、粒型、抗旱性、耐涝性、耐盐性、抗病性(稻瘟病、白叶枯病、褐斑病)
| QTL,GWAS | [32⇓-34] |
| | MAGIC PLUS | ― | 144 S8 | | | |
| | MAGIC PLUS
DH系 | ―
| 76 DH
| | | |
| 粳稻 | 8亲本 | Funnel杂交 | 500 S5 | | | |
| 籼―粳 | GLOBAL MAGIC | ― | 1402 S7 | | | |
小麦Wheat
| 春小麦
| 4亲本
| Funnel杂交
| 1579 F6
| 826 DArTs、283SNPs、53SSRs,9K SNP芯片,90K SNP芯片 | 株高、籽粒重、
胚芽鞘长度
| LMC,QTL
| [13,35-36]
|
| 冬小麦
| 8亲本
| Funnel杂交
| 1091 F7
| 90K SNP芯片
| 产量、开花期、
病害和株高 | LMC,QTL
| [37-38]
|
| 全麦粉
| 4亲本
| Funnel杂交
| 332 F6
| SSR
| 产量性状、
抗白粉病 | QTL,LMC
| [39]
|
| 欧洲
小麦 | 60亲本
| 随机杂交
| 1000 S4
| 9K SNP芯片
| 抽穗期
| LDA,GWAS
| [40]
|
| | | | | 14个KASPar SNP | | | |
大麦
Hordeum | – | 8亲本 | Funnel杂交 | 5000 DH | 9K SNP芯片 | 开花期 | QTL | [41] |
| 8亲本 | Funnel杂交 | 642 F5 | 9K SNP芯片 | 黄斑病 | LMC,QTL | [42] |
| 棉花Cotton | 陆地棉 | 11亲本 | 随机杂交 | 547 F5 | GBS | 纤维品质 | GWAS,QTL | [43] |
| | 8亲本
| 随机杂交
| 960 MLs
| SLAF-seq
| 开花期、株型、
果枝数 | GWAS,QTL
| [44]
|
| | 11亲本 | 随机杂交 | 550 RIL | 47K SNP芯片 | 株高、茎秆质量 | GWAS,QTL | [45] |
高粱
Sorghum | –
| 29亲本
| 随机杂交
| 1000 F6
| GBS
| 株高
| GWAS,QTL
| [46]
|
作物
Crop | 类型
Type | 亲本选择
Parental selection | 杂交方式
Hybridization | 群体规模
Group size | 基因型鉴定
Genotyping | 研究性状
Research trait | 分析方法
Analytical method | 参考文献
Reference |
烟草
Tobacco | –
| 8亲本
| 双列杂交
| 600 F4
| 430K 芯片
| 株型、烟叶品质
| GWAS
| [47]
|
玉米
Maize | –
| 8亲本
| Funnel杂交
| 1636 F6
| GBS
| 开花期、株高、
穗位高 | LMC,AM,QTL | [48]
|
| | 4亲本
| Funnel杂交
| 1149 S3
| 118K SNP 芯片
| 株高、穗高、散
粉期、吐丝期 | QTL,GWAS
| [49]
|
| | 8亲本
| Funnel杂交
| 401 RIL
| 56K SNP 芯片
| 苗重、苗长和
黄萎病 | QTL,NAM
| [50]
|
| | 8亲本
| Funnel杂交
| 700F6
| 110K SNP
芯片 | 镰孢菌穗腐病
| LMC,AM,QTL | [51]
|
| | 8亲本
| Funnel杂交
| 406 RIL
| 100K SNP
芯片,GBS | 发芽率、叶绿素
和出苗期 | GWAS,QTL
| [52]
|
| | 8亲本
| Funnel杂交
| 368 MLs
| 1000K SNP芯片,GBS | 籽粒含油量
| GWAS,QTL
| [53]
|
| | 16亲本
| Funnel杂交
| 513 RIL
| 500K SNP芯片
| 株高、叶形态、
开花期、穗大小 | GWAS
| [54]
|
| | 4亲本
| Funnel杂交
| 1044 F6
| 50K SNP芯片
| 产量、吐丝期、
株高 | QTL,AM
| [55]
|
油菜
Brassica napus | 甘蓝型
| 8亲本
| 随机杂交
| 680 F6
| –
| 始花期、抗病性
| SSD
| [56]
|
Fn表示构建F1的杂交过程和F1自交过程的总代数,Sn表示F1植株自交的代数,DH(doubled haploid)表示双单倍体系,GBS(genotyping by sequencing)表示基于测序的基因分型,QTL(quantitative trait locus)表示QTL定位,LDA(linkage disequilibrium analysis)表示连锁不平衡分析,LMC(linkage map construction)表示连锁图谱构建,AM(association mapping)表示关联作图,GWAS(genome wide association study)表示全基因组关联分析,SNP(single nucleotide polymorphisms)表示单核苷酸多态性,Funnel杂交表示“漏斗”型杂交,SSR(simple sequence repeat)表示简单重复序列,SSD(single seed descent)表示单粒传法,RIL(recombinant inbred lines)表示重组自交系,ML(lines)表示自交系,NAM(nested association map)表示巢式关联图谱 ...
Linkage analysis and association mapping qtl detection models for hybrids between multiparental populations from two heterotic groups:application to biomass production in maize(Zea mays L.)
1
2017
... Application of MAGIC group in plant genetics research
Table 1 作物
Crop | 类型
Type | 亲本选择
Parental selection | 杂交方式
Hybridization | 群体规模
Group size | 基因型鉴定
Genotyping | 研究性状
Research trait | 分析方法
Analytical method | 参考文献
Reference |
拟南芥
Arabidopsis | –
| 19亲本
| 随机杂交
| 1026 F4
| 1260 SNPs
| 抽薹期
| QTL
| [26]
|
| 水稻Rice | 籼稻 | 4亲本DC1 | 双列杂交 | 495 F6 | 6K SNP芯片 | 籽粒产量性状、穗相关性状、分蘖相关性状、抽穗期、苗期和株高
| GWAS | [23,28-31] |
| | 4亲本DC2 | 双列杂交 | 525 F6 | | | |
| | 8亲本
| Funnel杂交
| 668 F6
| 55K SNP芯片,
GBS | | |
| 籼稻 | 8亲本 | Funnel杂交 | 1328 S7 | GBS: 17387 SNP | 产量性状、稻米品质、开花期、株高、粒型、抗旱性、耐涝性、耐盐性、抗病性(稻瘟病、白叶枯病、褐斑病)
| QTL,GWAS | [32⇓-34] |
| | MAGIC PLUS | ― | 144 S8 | | | |
| | MAGIC PLUS
DH系 | ―
| 76 DH
| | | |
| 粳稻 | 8亲本 | Funnel杂交 | 500 S5 | | | |
| 籼―粳 | GLOBAL MAGIC | ― | 1402 S7 | | | |
小麦Wheat
| 春小麦
| 4亲本
| Funnel杂交
| 1579 F6
| 826 DArTs、283SNPs、53SSRs,9K SNP芯片,90K SNP芯片 | 株高、籽粒重、
胚芽鞘长度
| LMC,QTL
| [13,35-36]
|
| 冬小麦
| 8亲本
| Funnel杂交
| 1091 F7
| 90K SNP芯片
| 产量、开花期、
病害和株高 | LMC,QTL
| [37-38]
|
| 全麦粉
| 4亲本
| Funnel杂交
| 332 F6
| SSR
| 产量性状、
抗白粉病 | QTL,LMC
| [39]
|
| 欧洲
小麦 | 60亲本
| 随机杂交
| 1000 S4
| 9K SNP芯片
| 抽穗期
| LDA,GWAS
| [40]
|
| | | | | 14个KASPar SNP | | | |
大麦
Hordeum | – | 8亲本 | Funnel杂交 | 5000 DH | 9K SNP芯片 | 开花期 | QTL | [41] |
| 8亲本 | Funnel杂交 | 642 F5 | 9K SNP芯片 | 黄斑病 | LMC,QTL | [42] |
| 棉花Cotton | 陆地棉 | 11亲本 | 随机杂交 | 547 F5 | GBS | 纤维品质 | GWAS,QTL | [43] |
| | 8亲本
| 随机杂交
| 960 MLs
| SLAF-seq
| 开花期、株型、
果枝数 | GWAS,QTL
| [44]
|
| | 11亲本 | 随机杂交 | 550 RIL | 47K SNP芯片 | 株高、茎秆质量 | GWAS,QTL | [45] |
高粱
Sorghum | –
| 29亲本
| 随机杂交
| 1000 F6
| GBS
| 株高
| GWAS,QTL
| [46]
|
作物
Crop | 类型
Type | 亲本选择
Parental selection | 杂交方式
Hybridization | 群体规模
Group size | 基因型鉴定
Genotyping | 研究性状
Research trait | 分析方法
Analytical method | 参考文献
Reference |
烟草
Tobacco | –
| 8亲本
| 双列杂交
| 600 F4
| 430K 芯片
| 株型、烟叶品质
| GWAS
| [47]
|
玉米
Maize | –
| 8亲本
| Funnel杂交
| 1636 F6
| GBS
| 开花期、株高、
穗位高 | LMC,AM,QTL | [48]
|
| | 4亲本
| Funnel杂交
| 1149 S3
| 118K SNP 芯片
| 株高、穗高、散
粉期、吐丝期 | QTL,GWAS
| [49]
|
| | 8亲本
| Funnel杂交
| 401 RIL
| 56K SNP 芯片
| 苗重、苗长和
黄萎病 | QTL,NAM
| [50]
|
| | 8亲本
| Funnel杂交
| 700F6
| 110K SNP
芯片 | 镰孢菌穗腐病
| LMC,AM,QTL | [51]
|
| | 8亲本
| Funnel杂交
| 406 RIL
| 100K SNP
芯片,GBS | 发芽率、叶绿素
和出苗期 | GWAS,QTL
| [52]
|
| | 8亲本
| Funnel杂交
| 368 MLs
| 1000K SNP芯片,GBS | 籽粒含油量
| GWAS,QTL
| [53]
|
| | 16亲本
| Funnel杂交
| 513 RIL
| 500K SNP芯片
| 株高、叶形态、
开花期、穗大小 | GWAS
| [54]
|
| | 4亲本
| Funnel杂交
| 1044 F6
| 50K SNP芯片
| 产量、吐丝期、
株高 | QTL,AM
| [55]
|
油菜
Brassica napus | 甘蓝型
| 8亲本
| 随机杂交
| 680 F6
| –
| 始花期、抗病性
| SSD
| [56]
|
Fn表示构建F1的杂交过程和F1自交过程的总代数,Sn表示F1植株自交的代数,DH(doubled haploid)表示双单倍体系,GBS(genotyping by sequencing)表示基于测序的基因分型,QTL(quantitative trait locus)表示QTL定位,LDA(linkage disequilibrium analysis)表示连锁不平衡分析,LMC(linkage map construction)表示连锁图谱构建,AM(association mapping)表示关联作图,GWAS(genome wide association study)表示全基因组关联分析,SNP(single nucleotide polymorphisms)表示单核苷酸多态性,Funnel杂交表示“漏斗”型杂交,SSR(simple sequence repeat)表示简单重复序列,SSD(single seed descent)表示单粒传法,RIL(recombinant inbred lines)表示重组自交系,ML(lines)表示自交系,NAM(nested association map)表示巢式关联图谱 ...
甘蓝型油菜MAGIC群体构建及其在遗传育种中的应用潜力
1
2017
... Application of MAGIC group in plant genetics research
Table 1 作物
Crop | 类型
Type | 亲本选择
Parental selection | 杂交方式
Hybridization | 群体规模
Group size | 基因型鉴定
Genotyping | 研究性状
Research trait | 分析方法
Analytical method | 参考文献
Reference |
拟南芥
Arabidopsis | –
| 19亲本
| 随机杂交
| 1026 F4
| 1260 SNPs
| 抽薹期
| QTL
| [26]
|
| 水稻Rice | 籼稻 | 4亲本DC1 | 双列杂交 | 495 F6 | 6K SNP芯片 | 籽粒产量性状、穗相关性状、分蘖相关性状、抽穗期、苗期和株高
| GWAS | [23,28-31] |
| | 4亲本DC2 | 双列杂交 | 525 F6 | | | |
| | 8亲本
| Funnel杂交
| 668 F6
| 55K SNP芯片,
GBS | | |
| 籼稻 | 8亲本 | Funnel杂交 | 1328 S7 | GBS: 17387 SNP | 产量性状、稻米品质、开花期、株高、粒型、抗旱性、耐涝性、耐盐性、抗病性(稻瘟病、白叶枯病、褐斑病)
| QTL,GWAS | [32⇓-34] |
| | MAGIC PLUS | ― | 144 S8 | | | |
| | MAGIC PLUS
DH系 | ―
| 76 DH
| | | |
| 粳稻 | 8亲本 | Funnel杂交 | 500 S5 | | | |
| 籼―粳 | GLOBAL MAGIC | ― | 1402 S7 | | | |
小麦Wheat
| 春小麦
| 4亲本
| Funnel杂交
| 1579 F6
| 826 DArTs、283SNPs、53SSRs,9K SNP芯片,90K SNP芯片 | 株高、籽粒重、
胚芽鞘长度
| LMC,QTL
| [13,35-36]
|
| 冬小麦
| 8亲本
| Funnel杂交
| 1091 F7
| 90K SNP芯片
| 产量、开花期、
病害和株高 | LMC,QTL
| [37-38]
|
| 全麦粉
| 4亲本
| Funnel杂交
| 332 F6
| SSR
| 产量性状、
抗白粉病 | QTL,LMC
| [39]
|
| 欧洲
小麦 | 60亲本
| 随机杂交
| 1000 S4
| 9K SNP芯片
| 抽穗期
| LDA,GWAS
| [40]
|
| | | | | 14个KASPar SNP | | | |
大麦
Hordeum | – | 8亲本 | Funnel杂交 | 5000 DH | 9K SNP芯片 | 开花期 | QTL | [41] |
| 8亲本 | Funnel杂交 | 642 F5 | 9K SNP芯片 | 黄斑病 | LMC,QTL | [42] |
| 棉花Cotton | 陆地棉 | 11亲本 | 随机杂交 | 547 F5 | GBS | 纤维品质 | GWAS,QTL | [43] |
| | 8亲本
| 随机杂交
| 960 MLs
| SLAF-seq
| 开花期、株型、
果枝数 | GWAS,QTL
| [44]
|
| | 11亲本 | 随机杂交 | 550 RIL | 47K SNP芯片 | 株高、茎秆质量 | GWAS,QTL | [45] |
高粱
Sorghum | –
| 29亲本
| 随机杂交
| 1000 F6
| GBS
| 株高
| GWAS,QTL
| [46]
|
作物
Crop | 类型
Type | 亲本选择
Parental selection | 杂交方式
Hybridization | 群体规模
Group size | 基因型鉴定
Genotyping | 研究性状
Research trait | 分析方法
Analytical method | 参考文献
Reference |
烟草
Tobacco | –
| 8亲本
| 双列杂交
| 600 F4
| 430K 芯片
| 株型、烟叶品质
| GWAS
| [47]
|
玉米
Maize | –
| 8亲本
| Funnel杂交
| 1636 F6
| GBS
| 开花期、株高、
穗位高 | LMC,AM,QTL | [48]
|
| | 4亲本
| Funnel杂交
| 1149 S3
| 118K SNP 芯片
| 株高、穗高、散
粉期、吐丝期 | QTL,GWAS
| [49]
|
| | 8亲本
| Funnel杂交
| 401 RIL
| 56K SNP 芯片
| 苗重、苗长和
黄萎病 | QTL,NAM
| [50]
|
| | 8亲本
| Funnel杂交
| 700F6
| 110K SNP
芯片 | 镰孢菌穗腐病
| LMC,AM,QTL | [51]
|
| | 8亲本
| Funnel杂交
| 406 RIL
| 100K SNP
芯片,GBS | 发芽率、叶绿素
和出苗期 | GWAS,QTL
| [52]
|
| | 8亲本
| Funnel杂交
| 368 MLs
| 1000K SNP芯片,GBS | 籽粒含油量
| GWAS,QTL
| [53]
|
| | 16亲本
| Funnel杂交
| 513 RIL
| 500K SNP芯片
| 株高、叶形态、
开花期、穗大小 | GWAS
| [54]
|
| | 4亲本
| Funnel杂交
| 1044 F6
| 50K SNP芯片
| 产量、吐丝期、
株高 | QTL,AM
| [55]
|
油菜
Brassica napus | 甘蓝型
| 8亲本
| 随机杂交
| 680 F6
| –
| 始花期、抗病性
| SSD
| [56]
|
Fn表示构建F1的杂交过程和F1自交过程的总代数,Sn表示F1植株自交的代数,DH(doubled haploid)表示双单倍体系,GBS(genotyping by sequencing)表示基于测序的基因分型,QTL(quantitative trait locus)表示QTL定位,LDA(linkage disequilibrium analysis)表示连锁不平衡分析,LMC(linkage map construction)表示连锁图谱构建,AM(association mapping)表示关联作图,GWAS(genome wide association study)表示全基因组关联分析,SNP(single nucleotide polymorphisms)表示单核苷酸多态性,Funnel杂交表示“漏斗”型杂交,SSR(simple sequence repeat)表示简单重复序列,SSD(single seed descent)表示单粒传法,RIL(recombinant inbred lines)表示重组自交系,ML(lines)表示自交系,NAM(nested association map)表示巢式关联图谱 ...
Methods for linkage disequilibrium mapping in crops
1
2007
... 小麦基因组结构复杂,对于MAGIC群体也进行了深入遗传研究.第1个小麦MAGIC群体是Huang等[35]使用4个澳洲春小麦作为亲本构建了包含1579个 RILs的MAGIC群体.利用1162个DArT(diversity arrays technology)、SNP和SSR(simple sequence repeat)标记鉴定了871个家系的基因型并且构建了遗传图谱,检测到9个株高和8个百粒重的QTL,其中有3个株高QTL和已知基因相邻.Mackay等[37,57]采用8个冬小麦亲本构建了1个包含1091个家系的MAGIC群体.通过使用Illumina平台的90 000 SNP小麦芯片鉴定720个家系和8个亲本的基因型,共获得62 543个SNP标记.他们利用SNP标记通过连锁不平衡分析了基因组重组.小麦MAGIC群体是数据收集中进展最快的群体,推动了小麦遗传分析方法的发展,无论是在连锁图谱构建领域,还是标记和性状的关联定位,都取得了很好的进展. ...
陆地棉SSR标记遗传多样性及其与农艺性状的关联分析
1
2014
... 在棉花遗传学研究中,Islam等[43]和郭志军等[58]发现陆地棉MAGIC群体通过11个不同品种随机交配5代陆地棉MAGIC群体,利用6071个SNP标记,鉴定到一个影响纤维质量的基因GhRBB1_A07.有的研究[44,59-60]为了探索陆地棉重要农艺性状的遗传基础,从960 MLs的8亲本MAGIC群体中选择1个包含372个株系的MAGIC群体子集.利用60 495个SNP和表型数据进行全基因组关联作图,鉴定出177个 SNP在多个环境下与9个农艺性状显著相关.在117个QTL中有3个QTL在多种环境中是稳定的,11个QTL被证明与多个性状相关联. ...
棉花MAGIC群体后代株系对缩节胺的敏感性研究
1
2018
... 在棉花遗传学研究中,Islam等[43]和郭志军等[58]发现陆地棉MAGIC群体通过11个不同品种随机交配5代陆地棉MAGIC群体,利用6071个SNP标记,鉴定到一个影响纤维质量的基因GhRBB1_A07.有的研究[44,59-60]为了探索陆地棉重要农艺性状的遗传基础,从960 MLs的8亲本MAGIC群体中选择1个包含372个株系的MAGIC群体子集.利用60 495个SNP和表型数据进行全基因组关联作图,鉴定出177个 SNP在多个环境下与9个农艺性状显著相关.在117个QTL中有3个QTL在多种环境中是稳定的,11个QTL被证明与多个性状相关联. ...
基于自然群体及MAGIC群体关联分析解析陆地棉重要农艺性状的遗传基础
1
2018
... 在棉花遗传学研究中,Islam等[43]和郭志军等[58]发现陆地棉MAGIC群体通过11个不同品种随机交配5代陆地棉MAGIC群体,利用6071个SNP标记,鉴定到一个影响纤维质量的基因GhRBB1_A07.有的研究[44,59-60]为了探索陆地棉重要农艺性状的遗传基础,从960 MLs的8亲本MAGIC群体中选择1个包含372个株系的MAGIC群体子集.利用60 495个SNP和表型数据进行全基因组关联作图,鉴定出177个 SNP在多个环境下与9个农艺性状显著相关.在117个QTL中有3个QTL在多种环境中是稳定的,11个QTL被证明与多个性状相关联. ...
Natural variation in crops:realized understanding,continuing promise
1
2021
... 作物在生长发育过程中非常容易受到逆境胁迫的影响,提高抗逆性可以减少对作物产量的影响.逆境胁迫主要包括高温或低温、干旱、高盐和营养限制等非生物胁迫,还有病原菌和虫害侵入等生物胁迫,这些胁迫会对作物的正常生长发育产生巨大的影响,从而导致作物产量显著下降[61]. ...
The genomes of Oryza sativa:a history of duplications
1
2005
... 水稻MAGIC群体被应用于分析多种表型,其中研究最多的是复杂性状,如产量、耐旱和抗病性等.Bandillo等[32]构建了4个多亲本的群体,包括8亲本的粳稻MAGIC群体,8亲本的籼稻MAGIC群体,8亲本的籼稻MAGIC plus群体和16亲本(8个粳稻亲本和8个籼稻亲本)的Global MAGIC群体,并利用这些群体对水稻的抗稻瘟病、抗白叶枯病、耐盐性和耐涝性的稻米品质进行了全基因组关联分析,成功定位到显著性相关位点,包括与耐涝相关的基因Sub1和与抗白叶枯病相关的基因Xa4和Xa5.陈天晓等[33]和Yu等[62]利用8个具有丰富多样性的优良种质为亲本构建了2个8亲本和2个4亲本MAGIC群体.在这3个MAGIC群体分别接种GD-V(强致病菌)和C2(弱致病菌)致病力不同的水稻白叶枯病菌,对水稻抗白叶枯病进行QTL定位研究和群体结构分析,在3个群体917个稳定株系中共得到3128个高质量的SNP位点. ...
Linkage disequilibrium with linkage analysis of multiline crosses reveals different multiallelic QTL for hybrid performance in the flint and dent heterotic groups for maize
1
2014
... MAGIC群体不断得到育种家的认可并且用于各种作物中,同时也不断的在证明它在作物中的应用价值和优势.MAGIC群体可以更好地确定复杂性状的基因组区间,模拟复杂环境的相互作用和更好地预测在不同背景下等位基因效应.在一个基因组参考序列不可用的物种中,这些群体可提供用于产生高密度遗传连锁图谱.Giraud等[63]和Consortium[64]认为多亲本群体设计结合亲本密集基因分型,结合连锁不平衡信息和连锁分析方法,可提高QTL定位研究的多样性和分辨率.现在已经证明了MAGIC群体在传统遗传学分析中的价值,可以进行连锁图谱的构建、高精确率的连锁分析和关联分析.但真正考验MAGIC群体的是能否长期使用,并且能否把这些结果扩展到最新的基因组领域进行初步鉴定,进行高通量的遗传研究.下面对MAGIC群体优势领域进行展望和讨论,这将为我们未来理解复杂数量性状特性提供非常有价值的参考(图3). ...
The genome architecture of the colaborative cross mouse genetic reference population
1
2012
... MAGIC群体不断得到育种家的认可并且用于各种作物中,同时也不断的在证明它在作物中的应用价值和优势.MAGIC群体可以更好地确定复杂性状的基因组区间,模拟复杂环境的相互作用和更好地预测在不同背景下等位基因效应.在一个基因组参考序列不可用的物种中,这些群体可提供用于产生高密度遗传连锁图谱.Giraud等[63]和Consortium[64]认为多亲本群体设计结合亲本密集基因分型,结合连锁不平衡信息和连锁分析方法,可提高QTL定位研究的多样性和分辨率.现在已经证明了MAGIC群体在传统遗传学分析中的价值,可以进行连锁图谱的构建、高精确率的连锁分析和关联分析.但真正考验MAGIC群体的是能否长期使用,并且能否把这些结果扩展到最新的基因组领域进行初步鉴定,进行高通量的遗传研究.下面对MAGIC群体优势领域进行展望和讨论,这将为我们未来理解复杂数量性状特性提供非常有价值的参考(图3). ...
Whole-genome QTL analysis for MAGIC
1
2014
... 对于MAGIC,多亲本全基因组QTL分析可以利用全基因组平均区间图谱(whole-genome average interval mapping,WGAIM或MPWGAIM)延伸到多变量的情况[65-66].MPWGAIM涉及到每个潜在的QTL位点和每个亲本遗传等位基因的概率,允许连锁图谱上的每个区间或每个标记上都有1个QTL,无论在间隔内还是在标记处,都可以对假设QTL大小随机效应进行显著性检验来确定QTL是否存在.而Scutari等[67]采用了另一种贝叶斯网络的方法,利用冬小麦MAGIC群体的数据来探索贝叶斯网络同时建模分析多个数量性状的框架.结果表明,这种方法相当于多元最优线性无偏预测(genetic best liear unbiased prediction,GBLUP),因为MAGIC群体非常低的群体结构和大的样本容量使预测模型具有一个理想的环境,因此,在预测性能上与单性状GBLUP具有竞争性.此外,Malosetti等[38]说他们的双亲群体多变量模型可以很容易地扩展到多亲本群体.虽然MPWGAIM和贝叶斯网络都已应用到小麦MAGIC数据,但双方重点不同,未进行直接比较.对于所有的大数据统计分析方法,由于运用了较复杂的模型,需要耗费大量的时间和计算资源. ...
Genetic diversity and population structure of elite cotton(Gossypium hirsutum L.) germplasm revealed by SSR markers
1
2015
... 对于MAGIC,多亲本全基因组QTL分析可以利用全基因组平均区间图谱(whole-genome average interval mapping,WGAIM或MPWGAIM)延伸到多变量的情况[65-66].MPWGAIM涉及到每个潜在的QTL位点和每个亲本遗传等位基因的概率,允许连锁图谱上的每个区间或每个标记上都有1个QTL,无论在间隔内还是在标记处,都可以对假设QTL大小随机效应进行显著性检验来确定QTL是否存在.而Scutari等[67]采用了另一种贝叶斯网络的方法,利用冬小麦MAGIC群体的数据来探索贝叶斯网络同时建模分析多个数量性状的框架.结果表明,这种方法相当于多元最优线性无偏预测(genetic best liear unbiased prediction,GBLUP),因为MAGIC群体非常低的群体结构和大的样本容量使预测模型具有一个理想的环境,因此,在预测性能上与单性状GBLUP具有竞争性.此外,Malosetti等[38]说他们的双亲群体多变量模型可以很容易地扩展到多亲本群体.虽然MPWGAIM和贝叶斯网络都已应用到小麦MAGIC数据,但双方重点不同,未进行直接比较.对于所有的大数据统计分析方法,由于运用了较复杂的模型,需要耗费大量的时间和计算资源. ...
Multiple quantitative trait analysis using bayesian networks
2
2014
... 对于MAGIC,多亲本全基因组QTL分析可以利用全基因组平均区间图谱(whole-genome average interval mapping,WGAIM或MPWGAIM)延伸到多变量的情况[65-66].MPWGAIM涉及到每个潜在的QTL位点和每个亲本遗传等位基因的概率,允许连锁图谱上的每个区间或每个标记上都有1个QTL,无论在间隔内还是在标记处,都可以对假设QTL大小随机效应进行显著性检验来确定QTL是否存在.而Scutari等[67]采用了另一种贝叶斯网络的方法,利用冬小麦MAGIC群体的数据来探索贝叶斯网络同时建模分析多个数量性状的框架.结果表明,这种方法相当于多元最优线性无偏预测(genetic best liear unbiased prediction,GBLUP),因为MAGIC群体非常低的群体结构和大的样本容量使预测模型具有一个理想的环境,因此,在预测性能上与单性状GBLUP具有竞争性.此外,Malosetti等[38]说他们的双亲群体多变量模型可以很容易地扩展到多亲本群体.虽然MPWGAIM和贝叶斯网络都已应用到小麦MAGIC数据,但双方重点不同,未进行直接比较.对于所有的大数据统计分析方法,由于运用了较复杂的模型,需要耗费大量的时间和计算资源. ...
... 作物耐逆性受环境影响比较大,上位性作用显著.MAGIC群体的主要优点之一是通过一代又一代的混合亲本基因组创造新的等位基因组合.然而,在MAGIC的上位性相互作用,即等位基因的非加性相互作用,迄今为止还很少被人探索.与传统的上位性基于一个给定性状的基因座之间的非独立性基因型值相比,Corbett-Detig等[68]认为上位性也应该是具备一个基因组性能,并以检测基因型比畸变(genotype ratio distortion,GRD)作为上位性的标志,并将这种方法应用于果蝇黑腹重组自交系,和拟南芥的MAGIC群体以及其他多亲本群体双等位基因的相互作用.为了进一步验证这种方法,在拟南芥和玉米的MAGIC群体中筛选GRD,在拟南芥和玉米中分别发现了7个和5个GRD实例.Huang等[69]研究表明,检测到的主效应QTL之间存在相互作用,全基因组扫描对于检测非主效基因座之间的上位性是有效的.事实上,Scutari等[67]表明贝叶斯网络方法的一个缺点是它捕捉更小的上位性效应的能力有限,其中有主效基因控制的位点能够被SNP捕捉. ...
Genetic incompatibilities are widespresd within species
1
2013
... 作物耐逆性受环境影响比较大,上位性作用显著.MAGIC群体的主要优点之一是通过一代又一代的混合亲本基因组创造新的等位基因组合.然而,在MAGIC的上位性相互作用,即等位基因的非加性相互作用,迄今为止还很少被人探索.与传统的上位性基于一个给定性状的基因座之间的非独立性基因型值相比,Corbett-Detig等[68]认为上位性也应该是具备一个基因组性能,并以检测基因型比畸变(genotype ratio distortion,GRD)作为上位性的标志,并将这种方法应用于果蝇黑腹重组自交系,和拟南芥的MAGIC群体以及其他多亲本群体双等位基因的相互作用.为了进一步验证这种方法,在拟南芥和玉米的MAGIC群体中筛选GRD,在拟南芥和玉米中分别发现了7个和5个GRD实例.Huang等[69]研究表明,检测到的主效应QTL之间存在相互作用,全基因组扫描对于检测非主效基因座之间的上位性是有效的.事实上,Scutari等[67]表明贝叶斯网络方法的一个缺点是它捕捉更小的上位性效应的能力有限,其中有主效基因控制的位点能够被SNP捕捉. ...
MAGIC populations in crops:current status and future prospects
2
2015
... 作物耐逆性受环境影响比较大,上位性作用显著.MAGIC群体的主要优点之一是通过一代又一代的混合亲本基因组创造新的等位基因组合.然而,在MAGIC的上位性相互作用,即等位基因的非加性相互作用,迄今为止还很少被人探索.与传统的上位性基于一个给定性状的基因座之间的非独立性基因型值相比,Corbett-Detig等[68]认为上位性也应该是具备一个基因组性能,并以检测基因型比畸变(genotype ratio distortion,GRD)作为上位性的标志,并将这种方法应用于果蝇黑腹重组自交系,和拟南芥的MAGIC群体以及其他多亲本群体双等位基因的相互作用.为了进一步验证这种方法,在拟南芥和玉米的MAGIC群体中筛选GRD,在拟南芥和玉米中分别发现了7个和5个GRD实例.Huang等[69]研究表明,检测到的主效应QTL之间存在相互作用,全基因组扫描对于检测非主效基因座之间的上位性是有效的.事实上,Scutari等[67]表明贝叶斯网络方法的一个缺点是它捕捉更小的上位性效应的能力有限,其中有主效基因控制的位点能够被SNP捕捉. ...
... MAGIC群体及其衍生物为发展不同背景下优良等位基因的新组合提供种质资源,从而推进育种计划的实施.首先,可以直接将基因组选择的杂交组合后代放在田间实际大环境下进行正向选择.Huang等[69]提出了多亲本世代轮回选择(multi-parent advanced generation recurrent selection,MAGReS)的育种方法(图4).这个方法结合了MAGIC群体大量重组近交系的优势,利用分子标记辅助轮回选择互交的优良株系,选择培育具有不同亲本优异等位基因的新株系.多亲本世代轮回选择株系高度多样化,可作为不同育种品系的供体,优异品系可直接作为商业栽培的新品种来源. ...
EpiGPU:exhaustive pairwise epistasis scans parallelized on consumer level graphics cards
1
2011
... 对MAGIC群体的上位性的研究有3个主要难题.第1个是计算难度增加,由于大多数群体遗传标记的数量增加,从几千到几十万,测试两两相互作用的数量相应增加了4个数量级.随着并行计算应用,穷举搜索(exhaustive search)方法在人类关联研究中应用,可以接受在多个亲本的MAGIC群体中对数量性状进行线性回归或双性状或数量性状的上位性高通量分析[70].第2个问题实际上是源于亲本等位基因的多样性.在比较2个等位基因谁对性状影响更强时,仍需要测试不同亲本效应之间的差异.第3个问题是分析效率,这个问题取决于前2个问题解决的程度,检测上位性相互作用所需的样本量可能比检测相似大小的主效应所需的样本量大得多[71].因此,MAGIC群体上位相互作用分析的最佳方法就是对一系列实际数据的建模进行模拟应用,进而决定下一步如何去分析研究. ...
High throughput analysis of epistasis in genome-wide association studies with BiForce
0
2013



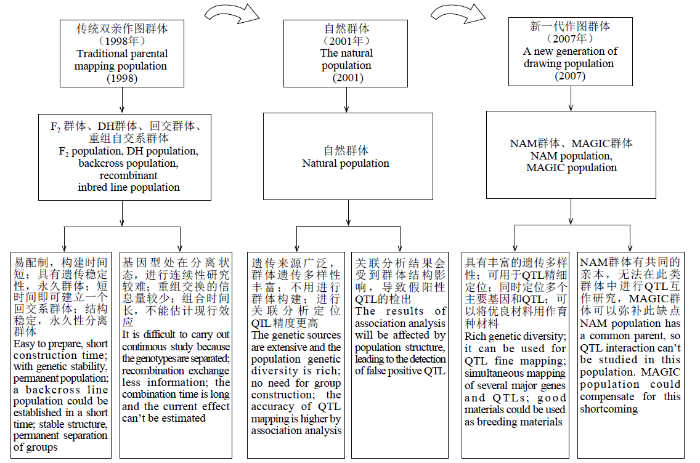
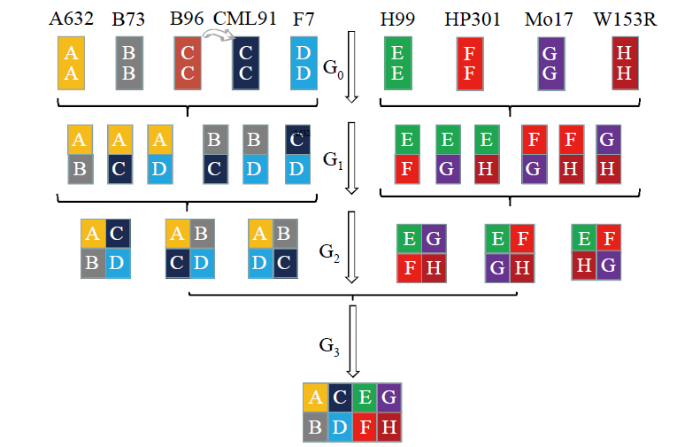
 "表示在F1 B96×HP301四元杂交构建失败,又引入第9亲本CML91代替B96作为B73×CML91双向杂交
"表示在F1 B96×HP301四元杂交构建失败,又引入第9亲本CML91代替B96作为B73×CML91双向杂交 "indicates that the construction of the quaternion hybrid B96×HP301 failed in F1 generation, and the ninth parent CML91 was introduced instead of B96 as a two-way cross of B73×CML91
"indicates that the construction of the quaternion hybrid B96×HP301 failed in F1 generation, and the ninth parent CML91 was introduced instead of B96 as a two-way cross of B73×CML91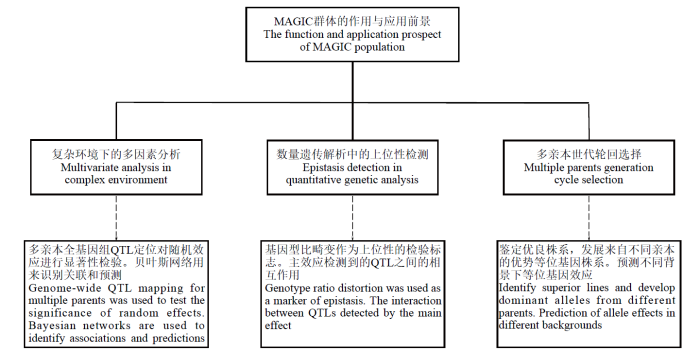
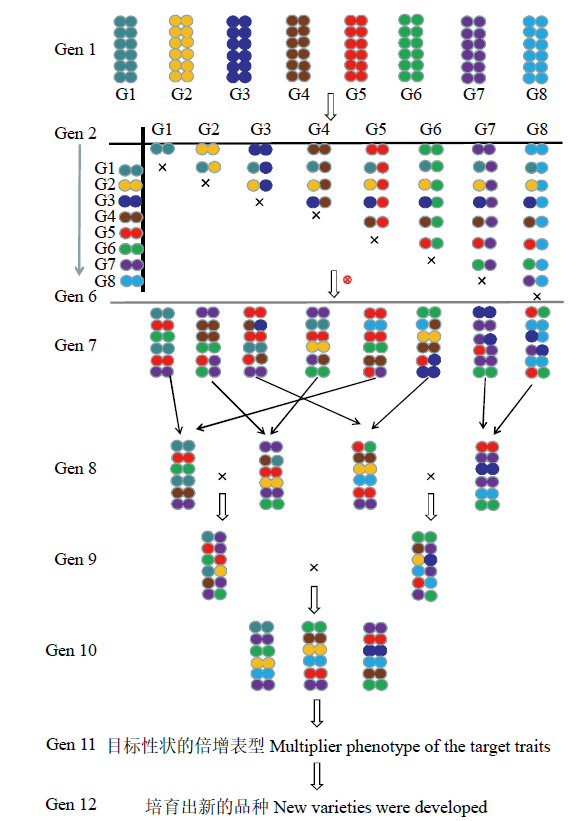


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}