


基因编辑技术的出现是生命科学发展的又一个里程碑,由于该技术操作简单和通用性强等优势,引起了科学各界的广泛关注,近几年的相关研究迅速增多[1]。2012年,Jennifer Doudna团队利用常间回文重复序列丛集/常间回文重复序列丛集关联蛋白系统(clustered regularly interspaced palindromic repeats/CRISPR-associated proteins system,CRISPR/Cas)实现了基因编辑[2];2013年,有研究人员通过CRISPR技术成功对真核细胞基因组进行编辑[3]。此后,该技术被广泛应用于植物、动物、微生物及人类疾病等众多领域,2020年法国科学家Emmanuelle Charpentier与美国科学家Jennifer A.Doudna也因“开发出一种基因编辑方法”而获得了诺贝尔化学奖。在此之前,测序技术的发展使得人们“读”取基因序列的能力提高,近几年随着基因编辑技术的极速发展使得人们开启了“写”的能力,该技术可以对基因组序列或基因转录产物进行有目的性的编辑[4]。本文对迄今为止各种基因编辑技术的原理及技术方法加以阐述,重点对商业化作物进展及检测研究加以详细分析,为未来海关进出口监管、检测、市场应用和安全评价等提供理论依据,为海关即将开展的相关科研课题研究以及标准制定提供参考。
1 基因编辑技术方法
基因编辑不同于转基因,它可实现基因的定向改造,即对特定的DNA片段进行添加和删除,及特定碱基的删除和替换等,从而达到对目的基因及产物的编辑,进而改变目标基因或调控元件的序列、结构或功能。
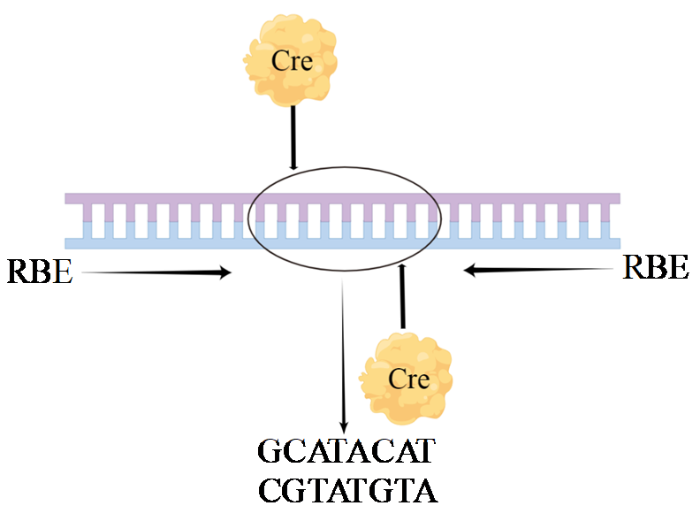
1.1 环化重组酶识别的回文DNA位点(cyclization recombinase locus of X-over P1,Cre-loxP)
目前该技术较多应用于微生物及植物领域,其方法来源于大肠杆菌(Escherichia coli,E.coli)P1噬菌体,可实现特异基因位点的插入、删除和易位等。来源于P1噬菌体的Cre重组酶及loxP识别位点构成了该系统。Cre酶能够识别基因中loxP位点并使其发生重组,从而实现定向编辑(图1)。该技术方法主要的重组方式是2个loxP位点在相同的DNA上,方向相同(敲除loxP序列)或相反(loxP序列翻转)以及2个loxP位点在不同的DNA上(交换或易位)。该方法简单、高效且特异性强,具有可控性及特异性[5]。缺点是只能在loxP位点特定位置进行插入和删除,不能对基因进行任意编辑,有局限性。
图1
1.2 锌指核酸内切酶(zinc figer nucleases,ZFNs)
核酸内切酶锌指(ZFNs)包含了切割存在于黄杆菌的IIS类型限制酶(IIS-type restriction enzymes present in Flavobacterium,Fok I)的结构域及重复锌指结构域的2个功能结构域的蛋白核酸内切酶。识别位点的特异性是由锌指结构决定的,因为每个锌指结构可以识别3个核酸。在目的位点两端都设计锌指酶,当Fok I形成二聚体后便将DNA双链断开,从而进行基因敲入、插入和删除等操作(图2)。锌指酶基因修饰具有高效定位和精确定位的优点,可以实现基因组的靶向敲除。缺点是锌指酶识别的目标序列长度是确定的,因此一些基因,包括一些小基因和同源性高的基因无法被敲除,大片段的基因很难被敲入。另外,潜在的脱靶效应是该方法最大的缺点,而且由于合成锌指酶的时间长,过程非模块化,所以该方法的应用受到了限制。
图2

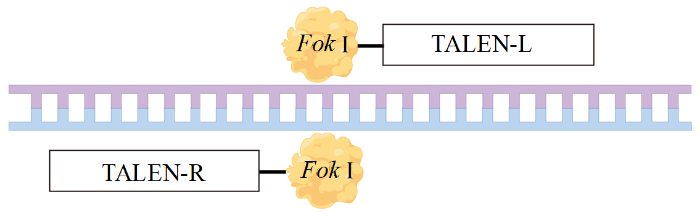
1.3 类转录激活因子效应物核酸酶(transcription activator-like effector nucleases,TALENs)定向修饰靶基因
TALENs是由类转录激活因子效应因子(TALEs)及Fok I的催化区域融合而成。TALENs与ZFNs的性质相似,通过识别特异的基因序列,并将其切割形成1个缺口,通过同源修复或非同源末端连接修复,从而导致基因插入或缺失[6](图3)。TALEs中的12和13位2个相邻的氨基酸被称为重复的变量二元结构(repeat variable di-residues,RVD),它们决定识别的位点。天冬酰胺与异亮氨酸可以识别A,天冬氨酸与组氨酸可以识别C,天冬酰胺与甘氨酸可以识别T,2个天冬酰胺则可以识别G。与ZFNs相比,TALENs易于操作,TALENs的识别位点范围更广,几乎可以敲除所有基因,所以TALENs的切割效率高于ZFNs。
图3
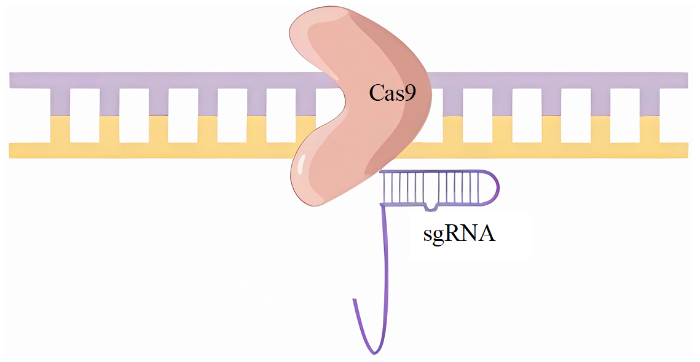
1.4 CRISPR/Cas9系统
CRISPR/Cas9系统中的Cas9与限制性核酸内切酶一样,可以切断DNA,不同之处在于,Cas9对DNA的识别需要小向导RNA(small guide RNA,sgRNA)与目标DNA配对,而切割则需要目标DNA上的前间隔序列邻近基序(protospacer adjacent motif,PAM)序列,也可以把Cas9的切割结构域融合表达成胞嘧啶脱氨酶之类的碱基,进而改造酶类,由此便可实现碱基的定点替换(图4)[7]。因此,想编辑某个基因,首先必须选择该基因上的PAM位点作为靶标,然后设计sgRNA在细胞中表达sgRNA和Cas9,切割靶标片段,形成一个双链缺口,通过同源修复或非同源末端连接进行修复,导致基因插入或缺失。该方法优点是可实现定点编辑,一次敲除多个基因,适用范围广,使用简单,成本低。缺点是切割受到PAM限制,偶尔会发生脱靶现象。
图4
1.5 Cas12a高效基因编辑系统
2021年10月13日,北京大学药学院天然药物及仿生药物国家重点实验室在《Molecular Cell》上发表了最新研究成果[8]。CRISPR/Cas12a相较于目前最为常用的CRISPR/Cas9系统具有更低的脱靶率和不同的识别序列,是一种更为精确的基因编辑系统,该编辑系统由蛋白质和核酸2个部分组成,核酸酶Cas12a蛋白负责切割基因组DNA,向导RNA(CRISPR-derived RNA,crRNA)则通过碱基互补配对的方式引导核酸酶到指定基因组位点进行切割。然而,由于Cas12a系统的基因编辑效率较低,严重限制了其应用发展。针对这一科学问题,有团队提出蛋白质核酸共价偶联复合物的策略,既通过生物正交反应实现Cas12a与crRNA的共价交联,从而将基因编辑中Cas12a、crRNA与基因组DNA之间的三分子反应转化为两分子反应,实现编辑效率的提升。该研究为Cas12a系统的广泛应用奠定了基础,并为其他低效的Cas核酸酶改造提供了思路与方法。
1.6 反转录子库重组工程(reverse transcription library recombination project,RLR)
1.7 基因编辑新工具:迷你CRISPR系统(mini CRISPR system,CasMINI)
2021年9月,《Molecular Cell》杂志上发表的一项研究[10]表明,一种名为CasMINI的紧凑高效的CRISPR/Cas系统可广泛用于细胞工程和基因治疗,因为它更容易进入细胞。CRISPR/Cas系统虽然优点多、应用广,但由于尺寸太大限制了其在细胞、组织或生物体的递送,进而阻碍了其临床应用。Cas12f被设计后解决了这一问题,它更高效、更紧凑,其大小在400~700氨基酸之间,不到目前使用的CRISPR系统的一半,比如Cas9或Cas12a。研究人员将RNA与蛋白工程应用到Cas12f系统,通过优化及筛选最终生成了一种Cas12f变体,称为CasMINI。CasMINI的分子大小仅为529个氨基酸,其应用更广泛。此外,可以将CasMINI mRNA包装到脂质纳米颗粒或其他RNA递送方式中,增强其进入细胞的能力,表现出有效的基因调控和基因编辑活性[10]。
1.8 光控CRISPR技术
CRISPR/Cas系统虽有众多优点,但在时间及空间上的可控性较差[11]。光调控已被证明是一种较为理想的非侵入式调控方法,将其引入到CRISPR/Cas系统中能够有效地改善CRISPR基因编辑技术中存在的脱靶性和潜在致癌性等问题[12⇓⇓⇓-16]。此技术使基因编辑可以进行时空调控,为该技术更广泛的应用开辟了新道路[13⇓-15]。该系统主要包括:(1)基于CRISPR/Cas系统的光学控制,该方法通过给CRISPR/Cas系统加上光控制“开关”,当有光照射时,系统便开始“工作”,降低了脱靶性,从而对基因编辑系统进行非常精确的控制[17⇓⇓-20]。目前,该方法主要在Cas蛋白、引导RNA以及递送系统的光诱导来实现对CRISPR/Cas系统的光操控,未来有望应用于基因功能的研究领域,以及遗传病、肿瘤等疾病的精确可控治疗[18⇓⇓⇓-22];(2)光诱导CRISPR/Cas9和CRISPR/Cas13系统,Pan等[13]将CRISPR/Cas9通过光敏分子“可光解的4-(羟甲基)-3-硝基苯甲酸”[photocleavable 4- (hydroxymethyl)-3-nitrobenzoic acid,ONA]锚定在上转换纳米材料(upconversion nanoparticles,UCNPs)上,然后用聚乙烯亚胺(PEI)进行包被,形成纳米颗粒。在经近红外光照射后,UCNPs转换,将光敏分子切断,CRISPR/Cas9从纳米颗粒中被释放出来,并将它们按需递送至细胞,实现靶向性。
近年来,已有多种基于CRISPR/Cas9系统和CRISPR/Cas13系统的光调控工具被开发,并已广泛应用于生物工程、基础医学和疾病治疗等领域。但光控CRISPR技术目前仍存在许多问题尚未解决,包括光毒性、光漂白、脱靶率、未知的安全隐患、递送效率、免疫原性和伦理问题等。
2 基因编辑应用及商业化
2.1 基因编辑应用
基因编辑技术已在疾病治疗领域表现出较强的优势,相关研究实现了对白血病、视网膜色素变性、杜氏肌营养不良以及心脏病等多种疾病的有效干预,并实现了特异性靶向敲除HIV病毒基因以及癌症融合基因,推动了疾病研究进程,同时还通过与嵌合抗原受体T细胞免疫疗法(Chimeric Antigen Receptor T-Cell Immunotherapy,CAR-T)、干细胞等先进生物技术的联用,助力再生医学疗法、基因疗法和癌症免疫疗法的开发和升级;另外,通过敲除猪基因组中所有可能的有害病毒基因,还将助力解决用猪器官进行人体移植中的重大难关[26]。在肿瘤[41]、直肠癌[42]、宫颈癌[43]和乳腺癌[44]等治疗研究方面也有进展。
利用先进的基因编辑技术对工程菌中复杂的代谢途径进行改造,可高效提升重组工程菌中目的产物的产量[45]。CRISPR/Cas9基因编辑系统已在放线菌[46]、食药用真菌[47]、念球菌[48]和微生物细胞[49]等方面得到研究应用。基因编辑技术极大提高了现代工业微生物育种的效率,TALENs和CRISPR技术相继应用在酿酒酵母、米曲霉菌、黑曲霉、谷氨酸棒状杆菌和产甘油假丝酵母等工业微生物育种中[50]。采用基因编辑技术构建的高效微生物工程菌,能够在保持菌体生长正常、代谢稳定的基础上提高已有工业微生物的产能和生理性能,缩短工程菌定向进化周期,提高突变体筛选效率,可以迅速提升产品多样性,是未来实现大规模生产的重要方向[45]。
2.2 基因编辑工具及作物商业化
2.2.1 基因编辑常用工具
基因编辑技术是一项重要的前沿新兴技术,已在生命科学理论研究、农作物的遗传改良以及人类疾病等领域得到广泛应用,掀起了一场颠覆性的革命[59]。基因编辑技术中最为典型及常用的工具为CRISPR、TALENs和ZFNs。各种工具的具体研究及应用现状如下。
(1)CRISPR:目前CRISPR应用的作物有阿拉伯芥、烟草、水稻和小麦,其中水稻和小麦主要以开发新抗病或耐逆境品种为主要研究方向,例如抗白叶枯病水稻、抗白粉病小麦等,其他作物还有高粱、玉米、番茄、柑橘、地钱和大豆等,目前已有提高糯玉米支链淀粉含量的基因编辑品种上市。除此之外,CRISPR被应用于真菌类上,例如抗褐化的蘑菇,目前有许多厂商正积极开发基因编辑作物,并且已有相关产品通过认定上市,如使用CRISPR/Cas9技术培育的高γ-氨基丁酸(gamma- aminobutyric acid,GABA)番茄品种已经被批准在日本种植和出售[60-61]。截至目前,利用该技术研究作物育种相关论文达篇1.3万余篇,专利5343个,涉及作物物种几十种。
(2)TALENs:目前TALENs应用的作物包括水稻、小麦、番茄、大豆和马铃薯等,例如抗白叶枯病水稻培育,番茄生长激素调控,马铃薯中已有降低褐变速率及减少丙烯酰胺产生的品种上市[60]。截至目前,利用该技术研究作物育种相关论文达800余篇,专利262个,涉及作物物种也比较广泛。
(3)ZFNs:目前ZFNs-1应用以烟草为主,另外也有使用抗除草剂使乙酰乳酸合成酶(acetolactate synthase,ALS)基因突变或带有筛选目标的β葡萄糖醛酸酶基因(beta glucuronidase gene,GUS)或绿色荧光蛋白(green fluorescent protein,GFP)植株植株;ZFNs-2主要应用于阿拉伯芥及带有突变基因GUS植株[60]。截至目前,利用该技术研究作物育种相关论文达410篇。到目前为止,国内外共有植物基因编辑育种企业124家,专利数达1782件。
2.2.2 商业化现状
2022年1月24日,中国农业农村部制定公布了《农业用基因编辑植物安全评价指南(试行)》,对于未引入外源基因的基因编辑植物,根据要求完成试验并提供相应安全数据资料的,可申请生产应用安全证书。受到这一政策的推动,一些基因编辑育种公司获得融资,并受到广泛关注。到目前为止,商业化的作物有以下几种,(1)使用CRISPR/Cas9技术培育的高GABA番茄品种已经被批准在日本种植和出售[61]。筑波大学植物分子生物学家Hiroshi Ezura说,GABA是日本著名的健康促进化合物,它就像维生素C,有助于降低血压并促进放松。(2)一种耐除草剂的油菜品种和利用TALENs基因编辑技术开发的具有高油酸低亚麻酸大豆品种在北美已商业化[62]。(3)韩国Toolgen公司于2021年初以来一直在中亚地区对利用CRISPR/Cas9改良的高油酸大豆进行田间试验,预计在未来3年内由小规模田间试种逐步扩大并实现商业化(数据来源于国际农业生物技术应用服务组织网站ISAAA INC.)[63]。(4)一种基因编辑的红鲷鱼“马代”于2021年10月在日本开始销售。用CRISPR基因编辑技术对该鱼的肌肉抑制素基因进行编辑,缺乏肌肉生长抑制素基因的红鲷鱼可食部分约增加至原有的1.2~1.6倍,饲料利用效率提高约14%(数据来源于ISAAA INC.)[64]。(5)2022年3月16日美国食品药品监督管理局(Food and Drug Administration,FDA)批准基因组编辑肉牛的营销。这是第一个针对食用动物基因组改变产品营销的决定。通过故意基因组改变(intentional genomic alteration,IGA)技术,即使用生物技术(包括基因组编辑)引入动物DNA的变化。肉牛中的IGA导致一些传统饲养的牛出现短毛皮毛特征,称为“光滑”皮毛。FDA通过对科学数据进行审查后,认为该产品为低风险产品,不会引起任何安全问题。因此,FDA决定开发商在产品上市前不需要提交批准申请(数据来源于ISAAA INC.)[65]。(6)比利时批准对基因编辑的玉米进行3项新的田间试验。获批的田间试验将与法兰德斯农业、渔业和食品研究所(Fisheries and Food,ILVO)密切合作进行,并且是比利时植物系统生物学中心研究项目的一部分。这些研究中的玉米是使用CRISPR/Cas9开发的,田间试验是由3位联邦卫生、环境和农业部长根据生物安全咨询委员会的赞成意见授权进行的(数据来源于ISAAA INC.)[66]。(7)其余已获得豁免权的作物有美国宾夕法尼亚大学的杨亦农用CRISPR/Cas9技术培育的基因编辑双孢蘑菇(Agaricus bisporus),通过直接敲除这种食用菌的1个表达多酚氧化酶的基因,可使其多酚氧化酶活性降低30%,从而不易发生酶促褐变;法国生物技术公司Cellectis的子公司Calyxt已经用TALENs技术培育出高油酸大豆、高油酸/低亚麻酸大豆、耐寒贮马铃薯、耐碰损马铃薯和品质改良苜蓿这些基因编辑作物品系[67]。
未来会有越来越多的基因编辑作物商业化,进入我国市场。但目前对于不同基因编辑技术商业化的作物检测方法未见,检验标准更是缺失,所以对于检测方法的研究是急需解决的重大问题。
3 基因编辑作物检测研究
由于基因编辑植物不同于传统的转基因生物,它不带有外源调控元件和功能基因,而是通过易错的非同源性末端接合途径或同源介导的双链DNA修复[68],修复DNA双链断裂会随机引入碱基插入、缺失或替换,这通常会导致基因插入、敲除或替换等基因组特定部位的突变。因此关于如何监管基因组编辑植物的讨论一直在进行,目前全球对基因编辑植物的监管没有明确规定。据日本广播公司NHK报道,日本政府的一个专家组决定,不对某些类型的编辑进行监管。美国农业部(United States Department of Agriculture,USDA)曾发表声明称,只要它们不是植物害虫,或是利用植物害虫开发的,就不会对利用基因组编辑技术改造过的植物进行监管。目前,美国对基因编辑的油菜、高油酸大豆、抗氧化蘑菇和糯玉米等作物均下达了转基因监管豁免权,将大多数基因编辑作物作为常规植物进行监管[69-70]。加拿大、哥伦比亚、巴西、阿根廷、智利、以色列和澳大利亚等国家监管政策与美国相同。欧洲最高法院裁定,基因编辑作物将面临与传统转基因生物同样的严格监管。欧洲法院认为,只要一种生物体的遗传物质发生了改变,并且不是通过交配或自然重组而发生的,就应被定义为转基因生物。挪威、尼日利亚、肯尼亚、巴拉圭和乌拉圭等国与欧洲法院相同,将进行逐案处理,如果没有外来DNA则不作为转基因生物进行监管。2022年1月24日,我国农业农村部制定公布了《农业用基因编辑植物安全评价指南(试行)》,对于未引入外源基因的基因编辑植物,根据要求完成试验并提供相应安全数据资料的,可申请生产应用安全证书,这一指南标志着我国将开始批准基因编辑作物。
无论各国政策如何,随着商业化进程发展和全球化贸易的形势,未来必将有基因编辑的作物进入国内市场,因此如何检验和监管成为急需解决的问题和挑战。已有研究人员针对检验方法进行研究,Zhang等[61]对基因编辑的水稻利用PCR及微滴数字PCR方法进行检测,并对盲样中0.1%~5.0%的基因编辑成分进行了准确定量,RSD<15%,偏差在±17%范围内,证明该方法满足检测方法的性能要求,为已知编辑序列的基因编辑植物提供了一种可靠的检测和定量方法。Peng等[71]通过实时定量PCR检测筛选了基因编辑的植物。随着基因编辑作物的商业化,如何对其进行准确检测和编制一套完善的标准检测体系已成为急需解决的问题和挑战[72]。
4 前景与展望
事物都有两面性,基因编辑技术也存在包括脱靶效应、染色体改变和潜在免疫原性的弊端,有研究[75]发现CRISPR有重大的潜在风险,在基因敲除过程中诱发的双链断裂可导致DNA产生损伤反应。美国国家癌症研究所研究发现,肿瘤抑制基因p53基因会识别DNA双链断裂并会阻止细胞分裂,p53是细胞生长周期中的负调节因子,p53基因突变的细胞通常是一些癌细胞。
此外,CRISPR基因编辑可能会给类似鼠类肉瘤病毒癌基因(kirsten rat sarcoma viral oncogene,KRAS)的致癌基因突变的细胞带来优势。脱靶效应也是不容忽视的重大问题,还有潜在的致癌问题。
5 总结
基因编辑技术问世以来取得了突飞猛进的发展,新技术、新方法、新研究和新成果层出不穷,这场技术风暴仍未停止。在未来,该技术将不断被开发改良并应用到众多领域中去,甚至有可能超越转基因成为植物改良的主要工具,同时会有越来越多的植物商业化,走出实验室,走进餐桌。因此,应当加速推进基因编辑植物的检测方法和检测标准的研究与制定,建立一套完善、精准、灵敏、高通量的基因编辑产品检测方法体系,用于促进基因编辑产品的进出口,保护我国农产品的经济和社会效益不受损坏,生态效益不受侵害,为我国生物安全监管提供有力的技术支撑。
参考文献
A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity
DOI:10.1126/science.1225829
PMID:22745249
[本文引用: 1]

Clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated (Cas) systems provide bacteria and archaea with adaptive immunity against viruses and plasmids by using CRISPR RNAs (crRNAs) to guide the silencing of invading nucleic acids. We show here that in a subset of these systems, the mature crRNA that is base-paired to trans-activating crRNA (tracrRNA) forms a two-RNA structure that directs the CRISPR-associated protein Cas9 to introduce double-stranded (ds) breaks in target DNA. At sites complementary to the crRNA-guide sequence, the Cas9 HNH nuclease domain cleaves the complementary strand, whereas the Cas9 RuvC-like domain cleaves the noncomplementary strand. The dual-tracrRNA:crRNA, when engineered as a single RNA chimera, also directs sequence-specific Cas9 dsDNA cleavage. Our study reveals a family of endonucleases that use dual-RNAs for site-specific DNA cleavage and highlights the potential to exploit the system for RNA-programmable genome editing.
Multiplex genome engineering using CRISPR/Cas systems
DOI:10.1126/science.1231143
PMID:23287718
[本文引用: 1]
Functional elucidation of causal genetic variants and elements requires precise genome editing technologies. The type II prokaryotic CRISPR (clustered regularly interspaced short palindromic repeats)/Cas adaptive immune system has been shown to facilitate RNA-guided site-specific DNA cleavage. We engineered two different type II CRISPR/Cas systems and demonstrate that Cas9 nucleases can be directed by short RNAs to induce precise cleavage at endogenous genomic loci in human and mouse cells. Cas9 can also be converted into a nicking enzyme to facilitate homology-directed repair with minimal mutagenic activity. Lastly, multiple guide sequences can be encoded into a single CRISPR array to enable simultaneous editing of several sites within the mammalian genome, demonstrating easy programmability and wide applicability of the RNA-guided nuclease technology.
Improving the efficiency of CRISPR-Cas12a-based genome editing with site-specific covalent Cas12a-crRNA conjugates
DOI:10.1016/j.molcel.2021.09.021
PMID:34648747
[本文引用: 1]
The CRISPR-Cas12a system shows unique features compared with widely used Cas9, making it an attractive and potentially more precise alternative. However, the adoption of this system has been hindered by its relatively low editing efficiency. Guided by physical chemical principles, we covalently conjugated 5' terminal modified CRISPR RNA (crRNA) to a site-specifically modified Cas12a through biorthogonal chemical reaction. The genome editing efficiency of the resulting conjugated Cas12a complex (cCas12a) was substantially higher than that of the wild-type complex. We also demonstrated that cCas12a could be used for precise gene knockin and multiplex gene editing in a chimeric antigen receptor T cell preparation with efficiency much higher than that of the wild-type system. Overall, our findings indicate that covalently linking Cas nuclease and crRNA is an effective approach to improve the Cas12a-based genome editing system and could potentially provide an insight into engineering other Cas family members with low efficiency as well.Copyright © 2021 Elsevier Inc. All rights reserved.
Engineered miniature CRISPR-Cas system for mammalian genome regulation and editing
DOI:10.1016/j.molcel.2021.08.008
PMID:34480847
[本文引用: 2]
Compact and versatile CRISPR-Cas systems will enable genome engineering applications through high-efficiency delivery in a wide variety of contexts. Here, we create an efficient miniature Cas system (CasMINI) engineered from the type V-F Cas12f (Cas14) system by guide RNA and protein engineering, which is less than half the size of currently used CRISPR systems (Cas9 or Cas12a). We demonstrate that CasMINI can drive high levels of gene activation (up to thousands-fold increases), while the natural Cas12f system fails to function in mammalian cells. We show that the CasMINI system has comparable activities to Cas12a for gene activation, is highly specific, and allows robust base editing and gene editing. We expect that CasMINI can be broadly useful for cell engineering and gene therapy applications ex vivo and in vivo.Copyright © 2021 Elsevier Inc. All rights reserved.
Genome Editing:past,present,and future
The CRISPR-Cas genome editing tools have been adopted rapidly in the research community, and they are quickly finding applications in the commercial sector as well. Lest we lose track of the broader context, this Perspective presents a brief review of the history of the genome editing platforms and considers a few current technological issues. It then takes a very limited view into the future of this technology and highlights some of the societal issues that require examination and discussion.
Near-infrared upconversion- activated CRISPR-Cas 9 system:a remote-controlled gene editing platform
DOI:10.1126/sciadv.aav7199
URL
[本文引用: 3]
Remote-controlled manipulation of gene editing was achieved in vivo.
Photoswitchable gRNAs for spatiotemporally controlled crisprcas- based genomic regulation
DOI:10.1021/acscentsci.9b01093
PMID:32490186
[本文引用: 2]
The recently discovered CRISPR-Cas gene editing system and its derivatives have found numerous applications in fundamental biology research and pharmaceutical sciences. The need for precise external control over the gene editing and regulatory events has driven the development of inducible CRISPR-Cas systems. While most of the light-controllable CRISPR-Cas systems are based on protein engineering, we developed an alternative synthetic approach based on modification of crRNA/tracrRNA duplex (guide RNA or gRNA) with photocaging groups, preventing the gRNA from recognizing its genome target sequence until its deprotection is induced within seconds of illumination. This approach relies on a straightforward solid-phase synthesis of the photocaged gRNAs, with simpler purification and characterization processes in comparison to engineering a light-responsive protein. We have demonstrated the feasibility of photocaging of gRNAs and light-mediated DNA cleavage upon brief exposure to light. We have achieved light-mediated spatiotemporally resolved gene editing as well as gene activation in cells, whereas photocaged gRNAs showed virtually no detectable gene editing or activation in the absence of light irradiation. Finally, we have applied this system to spatiotemporally control gene editing in zebrafish embryos, enabling the use of this strategy for developmental biology and tissue engineering applications.Copyright © 2020 American Chemical Society.
Optical control of a CRISPR/Cas 9 system for gene editing by using photolabile crRNA
Spatial and temporal control of CRISPR-Cas9-mediated gene editing delivered via a Light-Triggered liposome system
DOI:10.1021/acsami.0c16380 URL [本文引用: 1]
Photoactivatable CRISPR-Cas9 for optogenetic genome editing
DOI:10.1038/nbt.3245
PMID:26076431
[本文引用: 1]
We describe an engineered photoactivatable Cas9 (paCas9) that enables optogenetic control of CRISPR-Cas9 genome editing in human cells. paCas9 consists of split Cas9 fragments and photoinducible dimerization domains named Magnets. In response to blue light irradiation, paCas9 expressed in human embryonic kidney 293T cells induces targeted genome sequence modifications through both nonhomologous end joining and homology-directed repair pathways. Genome editing activity can be switched off simply by extinguishing the light. We also demonstrate activation of paCas9 in spatial patterns determined by the sites of irradiation. Optogenetic control of targeted genome editing should facilitate improved understanding of complex gene networks and could prove useful in biomedical applications.
Engineering a far-red light-activated split-Cas 9 system for remote-controlled genome editing of internal organs and tumors
Optical control of CRISPR/Cas 9 gene editing
DOI:10.1021/ja512664v
PMID:25905628
[本文引用: 2]
The CRISPR/Cas9 system has emerged as an important tool in biomedical research for a wide range of applications, with significant potential for genome engineering and gene therapy. In order to achieve conditional control of the CRISPR/Cas9 system, a genetically encoded light-activated Cas9 was engineered through the site-specific installation of a caged lysine amino acid. Several potential lysine residues were identified as viable caging sites that can be modified to optically control Cas9 function, as demonstrated through optical activation and deactivation of both exogenous and endogenous gene function.
Very fast CRISPR on damond
DOI:10.1126/science.aay8204
URL
[本文引用: 2]
\n Numerous efforts have been made to improve the temporal resolution of CRISPR-Cas9–mediated DNA cleavage to the hour time scale. Liu\n et al.\n developed a Cas9 system that achieved genome-editing manipulation at the second time scale (see the Perspective by Medhi and Jasin). Part of the guide RNA is chemically caged, allowing the Cas9-guide RNA complex to bind at a specific genomic locus without cleavage until activation by light. This fast CRISPR system achieves genome editing at high temporal resolution, enabling the study of early molecular events of DNA repair processes. This system also has high spatial resolution at short time scales, allowing editing of one genomic allele while leaving the other unperturbed.\n
Spatiotemporal control of CRISPR/Cas9 function in cells and zebrafish using light- activated guide RNA
The hidden land use cost of upscaling cover crops
CRISPR-Cas9 system:A new-fangled dawn in gene editing
DOI:10.1016/j.lfs.2019.116636 URL [本文引用: 1]
A noncanonical hippo pathway regulates spindle disassembly and cytokinesis during meiosis in saccharomyces cerevisiae
Dynamic temperature- sensitive A-to-I RNA editing in the brain of a heterothermic mammal during hibernation
DOI:10.1261/rna.066522.118
URL
[本文引用: 1]
RNA editing diversifies genomically encoded information to expand the complexity of the transcriptome. In ectothermic organisms, including Drosophila and Cephalopoda, where body temperature mirrors ambient temperature, decreases in environmental temperature lead to increases in A-to-I RNA editing and cause amino acid recoding events that are thought to be adaptive responses to temperature fluctuations. In contrast, endothermic mammals, including humans and mice, typically maintain a constant body temperature despite environmental changes. Here, A-to-I editing primarily targets repeat elements, rarely results in the recoding of amino acids, and plays a critical role in innate immune tolerance. Hibernating ground squirrels provide a unique opportunity to examine RNA editing in a heterothermic mammal whose body temperature varies over 30°C and can be maintained at 5°C for many days during torpor. We profiled the transcriptome in three brain regions at six physiological states to quantify RNA editing and determine whether cold-induced RNA editing modifies the transcriptome as a potential mechanism for neuroprotection at low temperature during hibernation. We identified 5165 A-to-I editing sites in 1205 genes with dynamically increased editing after prolonged cold exposure. The majority (99.6%) of the cold-increased editing sites are outside of previously annotated coding regions, 82.7% lie in SINE-derived repeats, and 12 sites are predicted to recode amino acids. Additionally, A-to-I editing frequencies increase with increasing cold-exposure, demonstrating that ADAR remains active during torpor. Our findings suggest that dynamic A-to-I editing at low body temperature may provide a neuroprotective mechanism to limit aberrant dsRNA accumulation during torpor in the mammalian hibernator.
番茄基因编辑研究进展和前景
DOI:10.16420/j.issn.0513-353x.2020-0976
[本文引用: 1]
介绍了基因编辑技术的发展和现状,并对该技术在番茄耐贮性和成熟调控、营养成分和果色、雄性不育和单性结实、逆境响应、株形、花形及野生种重新驯化等方面的研究进展进行了概述,梳理了基于CRISPR/Cas技术的单碱基编辑、DNA-free编辑及提高同源重组效率的技术创新及在番茄遗传改良和品种培育方面的应用前景和面临的挑战。
鱼类性别控制育种研究进展
DOI:10.13304/j.nykjdb.2021.0582
[本文引用: 1]
鱼类性别控制育种是水产遗传育种领域重要的研究方向之一。生殖内分泌调控、人工诱导雌核发育、种间杂交及分子标记辅助选育等技术被广泛用于养殖鱼类性别控制育种研究,已育成一批具有优良性状的单性养殖鱼类新品种。养殖鱼类基因组和功能基因组分析、性别决定与分化相关基因的发掘以及养殖鱼类高效特异基因编辑等前沿技术的建立,为养殖鱼类精准的性控育种新技术创建和新种质创制提供了重要的理论指导和技术支撑。概述了鱼类性别控制育种的理论基础以及性别控制技术的研究进展,以期为培育高产、优质和环境友好的单性养殖鱼类新品种提供理论指导及技术参考。
Targeted core-shell nanoparticles for precise CTCF gene insert in treatment of metastatic breast cancer
DOI:10.1016/j.bioactmat.2021.10.007 URL [本文引用: 1]
A systematic genome-wide mapping of oncogenic mutation selection during CRISPR-Cas 9 genome editing
DOI:10.1038/s41467-021-26788-6
[本文引用: 1]
Recent studies have reported that genome editing by CRISPR–Cas9 induces a DNA damage response mediated by p53 in primary cells hampering their growth. This could lead to a selection of cells with pre-existing p53 mutations. In this study, employing an integrated computational and experimental framework, we systematically investigated the possibility of selection of additional cancer driver mutations during CRISPR-Cas9 gene editing. We first confirm the previous findings of the selection for pre-existing p53 mutations by CRISPR-Cas9. We next demonstrate that similar to p53, wildtype KRAS may also hamper the growth of Cas9-edited cells, potentially conferring a selective advantage to pre-existing KRAS-mutant cells. These selective effects are widespread, extending across cell-types and methods of CRISPR-Cas9 delivery and the strength of selection depends on the sgRNA sequence and the gene being edited. The selection for pre-existing p53 or KRAS mutations may confound CRISPR-Cas9 screens in cancer cells and more importantly, calls for monitoring patients undergoing CRISPR-Cas9-based editing for clinical therapeutics for pre-existing p53 and KRAS mutations.
Highly sensitive and specific detection of hepatitis B virus DNA and drug resistance mutations utilizing the PCR-based CRISPR-Cas13a system
DOI:10.1016/j.cmi.2020.04.018 URL [本文引用: 1]
Colorimetric detection of SARS-CoV-2 and drug-resistant pH1N1 using CRISPR/dCas9
DOI:10.1021/acssensors.0c01929
PMID:33270431
[本文引用: 1]
Viruses have been a continuous threat to human beings. The coronavirus disease 2019 (COVID-19), caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), has led to a pandemic that is still ongoing worldwide. Previous pandemic influenza A virus (pH1N1) might be re-emerging through a drug-resistant mutation. We report a colorimetric viral detection method based on the clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 endonuclease dead (dCas9) system. In this method, RNA in the viral lysate was directly recognized by the CRISPR/dCas9 system with biotin-protospacer adjacent motif (PAM)-presenting oligonucleotide (PAMmer). Streptavidin-horseradish peroxidase then bound to biotin-PAMmer, inducing a color change through the oxidation of 3,3',5,5'-tetramethylbenzidine. Using the developed method, we successfully identified SARS-CoV-2, pH1N1, and pH1N1/H275Y viruses by the naked eye. Moreover, the detection of viruses in human nasopharyngeal aspirates and sputum was demonstrated. Finally, clinical samples from COVID-19 patients led to a successful diagnosis. We anticipate that the current method can be employed for simple and accurate diagnosis of viruses.
A novel CRISPR-based malaria diagnostic capable of plasmodium detection,speciation,and drug-resistance genotyping
DOI:10.1016/j.ebiom.2021.103415 URL [本文引用: 1]
Ultrasensitive CRISPR- based diagnostic for field-applicable detection of plasmodium species in symptomatic and asymptomatic malaria
Nucleic acid detection of plant genes using CRISPR-Cas13
DOI:10.1089/crispr.2019.0011
[本文引用: 1]
Nucleic acid detection is vital for agricultural applications including trait detection during breeding, pest surveillance, and pathogen identification. Here, we use a modified version of the CRISPR-based nucleic acid detection platform SHERLOCK to quantify levels of a glyphosate resistance gene in a mixture of soybeans and to detect multiple plant genes in a single reaction. SHERLOCK is rapid (similar to 15 min), quantitative, and portable, and can process crude soybean extracts as input material for minimal nucleic acid sample preparation. This field-ready SHERLOCK platform with color-based lateral flow readout can be applied for detection and quantitation of genes in a range of agricultural applications.
CRISPR-Cas12a-assisted multicolor biosensor for semiquantitative point-of-use testing of the nopaline synthase terminator in genetically modified crops by unaided eyes
DOI:10.1021/acssynbio.0c00365
PMID:33047952
[本文引用: 1]
Existing methods of detecting foreign genes and their expression products from genetically modified organisms (GMOs) suffer from the requirement of professional equipment and skillful operators. The same problem stays for the CRISPR-Cas12a system, although it has been emerging as a powerful tool for nucleic acid detection due to its remarkable sensitivity and specificity. In this report, a portable platform for the visible detection of GMOs based on CRISPR-Cas12a was established, which relies on a color change of gold nanorods (GNRs) caused by the invertase-glucose oxidase cascade reaction and the Fenton reaction for signal readout. A nopaline synthase (NOS) terminator was employed as a model target commonly existing in foreign genes of GMOs. With the help of recombinase-aided amplification, this platform achieved comparable sensitivity of DNA targets (1 aM) with that of a fluorescence reporting assay. As low as 0.1 wt % genetically modified (GM) content in Bt-11 maize was visually observed by unaided eyes, and the semiquantitation of GM ingredients can be obtained within the range of 0.1 to 40 wt % through the absorption measurement of GNRs. Furthermore, five real samples were tested by our method, and the results indicated that the GM ingredient percentages of GMO samples were 2.24 and 24.08 wt %, respectively, while the other three samples were GMO-free. With the advantages of a simple procedure, no need for large or professional instruments, high sensitivity, and selectivity, this platform is expected to provide reasonable technical support for the safe supervision of GMOs.
基于CRISPR/Cas原理的转基因产品检测技术研究进展
DOI:10.19586/j.2095-2341.2021.0083
[本文引用: 1]
随着转基因产品商业化种植面积不断增加、国际贸易日趋频繁,对转基因生物安全管理提出了更高的要求。转基因产品检测技术作为安全评价的关键环节,逐渐引起了各国政府的关注。目前,针对转基因产品的快速检测方法层出不穷,但这些检测方法对于设备、试剂和专业的实验人员均有较高的要求。因此,为了有效支撑转基因相关产业的发展和管理,亟需建立一种高灵敏度、高特异性及高效的转基因检测技术。基因组编辑技术是近年来迅速发展的一类遗传修饰技术,其代表技术——CRISPR/Cas技术,更是极大地推动了生物技术的发展。CRISPR/Cas技术除了被应用于基因编辑领域,也逐渐被应用于核酸分子检测领域。基于此,以转基因产品检测技术为立足点,从CRISPR/Cas的检测原理、检测效果等技术层面分析了CRISPR/Cas检测技术发展的必然性,并对其在转基因产品检测上的应用前景进行展望,旨在为我国转基因产品快速检测和有效监管工作提供资料,对于保障我国转基因产品贸易的顺利进行具有重要意义。
An editing-site-specific PCR method for detection and quantification of CAO1-edited rice
DOI:10.3390/foods10061209
URL
[本文引用: 3]
Genome-edited plants created by genome editing technology have been approved for commercialization. Due to molecular characteristics that differ from classic genetically modified organisms (GMOs), establishing regulation-compliant analytical methods for identification and quantification of genome-edited plants has always been regarded as a challenging task. An editing-site-specific PCR method was developed based on the unique edited sequence in CAO1-edited rice plants. Test results of seven primer/probe sets indicated that this method can identify specific CAO1-edited rice from other CAO1-edited rice and wild types of rice with high specificity and sensitivity. The use of LNA (locked nucleic acid) in a probe can efficiently increase the specificity of the editing-site-specific PCR method at increased annealing temperature which can eliminate non-specific amplification of the non-target. The genome-edited ingredient content in blinded samples at the level of 0.1% to 5.0% was accurately quantified by this method on the ddPCR platform with RSD of <15% and bias in the range of ±17%, meeting the performance requirements for GMO detection method. The developed editing-site-specific PCR method presents a promising detection and quantification technique for genome-edited plants with known edited sequence.
Real-time quantitative PCR method specific for detection and quantification of the first commercialized genome-edited plant
DOI:10.3390/foods9091245
URL
[本文引用: 1]
Discussion regarding the regulatory status of genome-edited crops has focused on precision of editing and on doubts regarding the feasibility of analytical monitoring compliant with existing GMO regulations. Effective detection methods are important, both for regulatory enforcement and traceability in case of biosafety, environmental or socio-economic impacts. Here, we approach the analysis question for the first time in the laboratory and report the successful development of a quantitative PCR detection method for the first commercialized genome-edited crop, a canola with a single base pair edit conferring herbicide tolerance. The method is highly sensitive and specific (quantification limit, 0.05%), compatible with the standards of practice, equipment and expertise typical in GMO laboratories, and readily integrable into their analytical workflows, including use of the matrix approach. The method, validated by an independent laboratory, meets all legal requirements for GMO analytical methods in jurisdictions such as the EU, is consistent with ISO17025 accreditation standards and has been placed in the public domain. Having developed a qPCR method for the most challenging class of genome edits, single-nucleotide variants, this research suggests that qPCR-based method development may be applicable to virtually any genome-edited organism. This advance resolves doubts regarding the feasibility of extending the regulatory approach currently employed for recombinant DNA-based GMOs to genome-edited organisms.
High-throughput detection and screening of plants modified by gene editing using quantitative real-time polymerase chain reaction
DOI:10.1111/tpj.13961
PMID:29761864
[本文引用: 1]
Gene editing techniques are becoming powerful tools for modifying target genes in organisms. Although several methods have been developed to detect gene-edited organisms, these techniques are time and labour intensive. Meanwhile, few studies have investigated high-throughput detection and screening strategies for plants modified by gene editing. In this study, we developed a simple, sensitive and high-throughput quantitative real-time (qPCR)-based method. The qPCR-based method exploits two differently labelled probes that are placed within one amplicon at the gene editing target site to simultaneously detect the wild-type and a gene-edited mutant. We showed that the qPCR-based method can accurately distinguish CRISPR/Cas9-induced mutants from the wild-type in several different plant species, such as Oryza sativa, Arabidopsis thaliana, Sorghum bicolor, and Zea mays. Moreover, the method can subsequently determine the mutation type by direct sequencing of the qPCR products of mutations due to gene editing. The qPCR-based method is also sufficiently sensitive to distinguish between heterozygous and homozygous mutations in T transgenic plants. In a 384-well plate format, the method enabled the simultaneous analysis of up to 128 samples in three replicates without handling the post-polymerase chain reaction (PCR) products. Thus, we propose that our method is an ideal choice for screening plants modified by gene editing from many candidates in T transgenic plants, which will be widely used in the area of plant gene editing.© 2018 The Authors The Plant Journal © 2018 John Wiley & Sons Ltd.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}