


目前,对玉米等作物脂肪酸合成相关基因的研究是分子育种工作的重点。Li等[6]基于368个不同的玉米品系,利用GWAS鉴定出与玉米油脂代谢途径相关的基因FAD2、LCACS、ACP、COPII和转录因子WRI1a。杨凤萍等[7]发现,双功能去饱和酶基因AcD12可以改良玉米不饱和脂肪酸成分,增加种子中亚油酸的含量。Shen等[8]发现ZmWRI1作为糖酵解和脂肪酸途径基因表达的关键转录因子,可以提高玉米的油脂产量。Wang等[9]通过GWAS分析,成功鉴定出与大豆亚油酸含量相关的SNP位点,并根据基因功能注释,挖掘出可能参与调节亚油酸积累的基因GmGA20ox。Di等[10]研究表明,转录因子GmWRI14触发FAD2基因下调是大豆种子亚油酸水平降低的潜在机制。此外,过表达miR319a改变了转基因拟南芥种子油的组成,导致亚油酸含量增加5.2%,油酸含量减少24.7%[11]。这些研究结果为亚油酸的合成与代谢提供了新的见解,并为进一步深入挖掘候选基因提供了理论依据。
近年来,转录组测序(RNA-seq)已被广泛应用于揭示复杂的生物学通路与分子调控机制中[12
(GDSL家族酯酶/脂肪酶)催化三酰甘油合成的最后一步,在2个高油型甜玉米中的表达水平显著高于2个非高油型甜玉米。Pouvreau等[18]研究表明,ZmWri1a基因的过表达增加了成熟玉米籽粒的脂肪酸含量,通过转录组分析鉴定出转录因子ZmWri1a调控18个假定靶基因。
本研究以自主育成的高亚油酸玉米自交系YM-2和普通玉米自交系昌7-2为材料,通过转录组学分析,研究DEGs的表达情况及转录调控机制,挖掘玉米响应亚油酸合成与代谢的关键通路和候选基因,以期为培育高亚油酸新品种提供理论依据。
1 材料与方法
1.1 试验材料
供试材料为自主育成的高亚油酸玉米自交系YM-2和普通玉米自交系昌7-2(C7-2),播种于江苏省农业科学院宿迁农科所。
1.2 测定项目与方法
1.2.1 农艺性状
于收获期,从YM-2和昌7-2中各随机选取15株植株,测定田间株高、穗数、穗位高、千粒重、穗型和株型等农艺性状。
1.2.2 亚油酸含量
取自然状态下生长的YM-2和昌7-2苗期叶片及新鲜籽粒,迅速置于液氮冷冻后,保存于-80 ℃冰箱备用,每个样品设置3次重复。取100 g样品置于100 mL比色管中,加入2 mL 95%乙醇和4 mL蒸馏水,再加入10 mL浓度为8.3 mol/L的盐酸溶液混合均匀,将盛有混合液的烧瓶置于80 ℃水浴中水解40 min,水解完成后取出样品冷却至室温;随后加入10 mL 95%乙醇,用100 mL乙醚/石油醚混合液(1:1)分3次提取,合并提取液至100 mL平底烧瓶中,将石油醚/乙醚层蒸干获取脂肪提取物;在脂肪提取物中加入4 mL2%氢氧化钠的甲醇溶液,于45 ℃水浴锅中水浴20 min,重复操作2次;随后在离心管中加入3 mL正己烷,震荡萃取2 min之后静置至分层,取上层清液,用0.45 μm滤膜过滤后采用气相色谱―气质联用仪(GC-MS)进行脂肪酸成分分析;通过MS得到其离子峰,根据质谱库对离子进行鉴定,同时利用软件MassHunter对化合物峰面积进行积分,定量分析脂肪酸。
1.3 转录组测序及质量评估
使用Illumina NovaSeq 6000平台对YM-2和昌7-2的苗期样品(共6份)总RNA进行有参转录组测序。将下机数据进行质控过滤,将得到的连续碱基序列(Reads)与参考基因组进行比对。将原始序列Reads归一化,选择文库中3个独立生物重复的平均归一化Reads用于后续分析。
1.4 样品间相关性分析
FPKM密度分布体现各样本的蛋白编码基因表达模式。根据转录本定量结果,使用R语言计算样品间相关性。通过样本间相关系数热图,评估组内生物学重复效果和组间样本差异。相关系数接近于1则表明两两样品间相似度高、差异小。
1.5 差异表达分析
利用DESeq2软件对各个样本基因的Counts数目进行标准化处理,计算差异倍数,并采用负二项分布进行差异显著性检验,最终根据差异倍数及差异显著性检验结果来筛选差异蛋白编码基因。设定显著性DEGs的阈值为q值<0.05、|log2FC|>1,挖掘YM-2和昌7-2样本间存在的DEGs。
1.6 GO富集和KEGG富集分析
使用生物生信在线工具平台(
1.7 玉米叶片RNA的提取及qRT-PCR验证
玉米叶片RNA提取所用的枪头为Axygen的10 μL、200 μL和1 mL无RNA无DNA酶的灭菌吸头;离心管为Axygen的1.5 mL和2.0 mL无DNA酶和RNA酶的灭菌管;研钵提前用氯仿浸泡5 min;RNAiso Plus、氯仿和无水乙醇均为提取RNA专用;RNA吸附与沉淀所用的试剂盒为EasyPure RNA Purification Kit(北京全式金生物技术股份有限公司,ER701),按步骤进行RNA提取。用琼脂糖凝胶电泳检测RNA是否降解,用NanoDrop 2000c微量分光光度计测定RNA浓度。剩余的RNA用于cDNA合成,进行qRT-PCR。
qRT-PCR反应在CFX96 Touch Real-Time PCR Detection System(Bio-Rad)仪器上进行,定量反应的试剂为Universal SYBR Green Supermix。通常将cDNA用ddH2O稀释10倍,按以下操作进行qRT-PCR,反应体系为Universal SYBR Green Supermix 4 μL、引物(5 mmol/L)0.6 μL、cDNA 0.5 μL、H2O(RNase Free)2.9 μL。反应条件为95 ℃预变性3 min;95 ℃变性10 s,50~65 ℃退火,延伸10~60 s,重复39个循环;65~95 ℃,每步增加0.5 ℃,5 s(建立溶解曲线确定扩增产物特异性)。用2-ΔΔCT法计算基因的相对表达量。内参基因为玉米GAPDH,引物信息见表1。
表1 qRT-PCR所用引物序列
Table 1
| 基因Gene | 上游引物序列Upstream primer sequence | 下游引物序列Downstream primer sequence |
|---|---|---|
| LOX3 | AGATGCAGGCCGTGTC | GGAGTGCTTGGAGAGG |
| LOX10 | AACGAGCACCACAAGGAGG | TCGGACGGCAGGTATGACT |
| TS1 | ACGGGAAGAACGAGAT | CCGCAGCCAAGCAAAC |
| LOC103642039 | GTCGTGCCTCGCCTGCTC | TGATTCCGCCTGTCCGTA |
2 结果与分析
2.1 YM-2和昌7-2的亚油酸含量及田间农艺性状
YM-2和昌7-2苗期叶片和新鲜籽粒的亚油酸含量测定结果(图1)表明,YM-2的新鲜籽粒亚油酸含量比昌7-2多89.9%,YM-2的苗期叶片亚油酸含量比昌7-2多12.7%。农艺性状考察结果发现,昌7-2的株高显著高于YM-2,多数植株高度差值在10 cm以上;而YM-2的千粒重显著大于昌7-2。YM-2穗位高在65 cm左右,穗位高与株高较为协调,穗型为圆筒形,穗数明显低于昌7-2,株型以半紧凑为主;昌7-2穗位高约为85.7 cm,穗型为短锥形,株型多为紧凑型。
图1
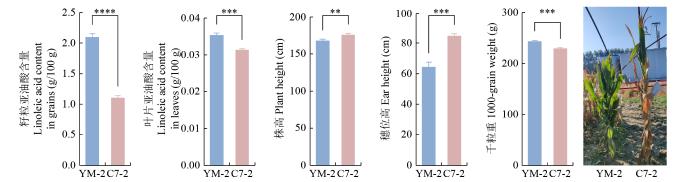
图1
YM-2和昌7-2的亚油酸含量及田间农艺性状
“**”、“***”和“****”分别代表同一比较组中YM-2和昌7-2在P < 0.01、P < 0.001和P < 0.0001水平上差异显著。下同。
Fig.1
Linoleic acid content and field agronomic traits of YM-2 and C7-2
“**”,“***”and“****”indicate that YM-2 and C7-2 in the same comparison group were significantly different at P < 0.01, P < 0.001, and P < 0.0001 levels, respectively. The same below.
2.2 YM-2和昌7-2苗期样品的转录组测序数据质量评估
为探究亚油酸高产机制,筛选亚油酸积累相关基因及通路途径,对6个样本进行转录组测序,经质控过滤后共获得了41.13 Gb的有效数据量,各组样本的过滤后数据(clean data)在6.39~6.97 Gb,Q30碱基质量值在95.65%~95.93%,平均GC含量为55.68%。同时,将得到的Reads与参考基因组比对,各样本的基因组比对率为90.35%~91.70%。以上结果均表明测序数据质量良好,可用于后续样品间DEGs比较分析。
2.3 YM-2和昌7-2间DEGs表达分析
图2
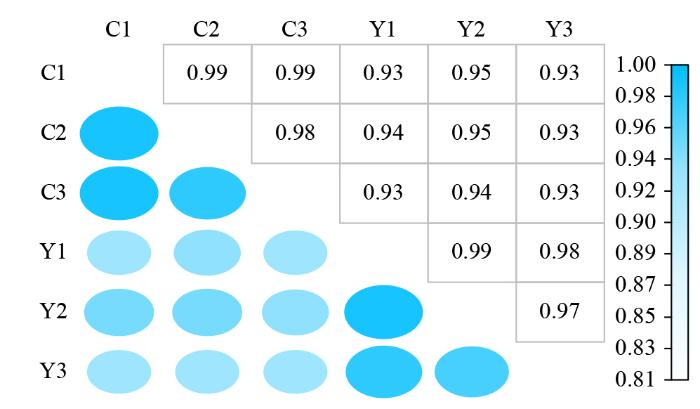
图3
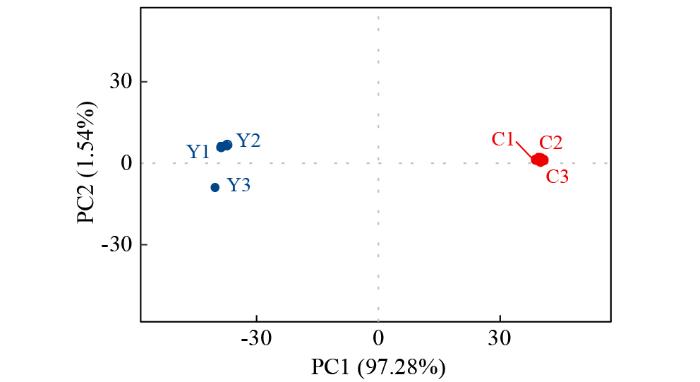
图4
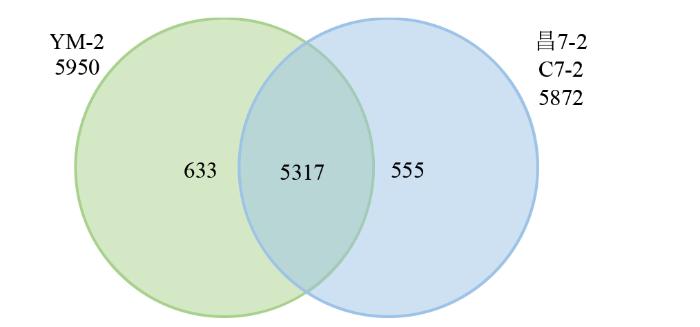
图4
YM-2和昌7-2样本中差异表达的基因
Fig.4
The differentially expressed genes in YM-2 and C7-2
图5
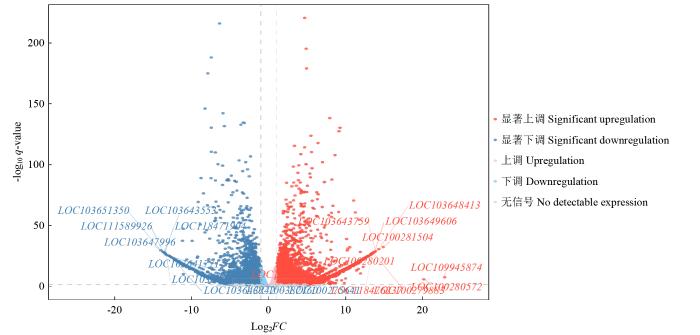
2.4 YM-2和昌7-2间DEGs的GO分析
对YM-2与昌7-2间的DEGs进行GeneOntology(GO)富集分析,GO term共分为3个模块,分别描述基因的分子功能(molecular function,MF)、细胞组分(cellular component,CC)和生物过程(biological process,BP),被分为包含生物黏附性(biological adhesion)、细胞性(cell)和结合性(binding)等的64个条目。其中,6505个DEGs主要富集到植物型初生细胞壁生物发生、质膜的整体成分和纤维素合成酶活性等条目中,在YM-2中上调表达的3381个基因主要富集到谷胱甘肽代谢过程、细胞器膜和铁离子结合等条目中,在YM-2中下调表达的3124个基因主要富集到植物型初生细胞壁生物发生、质膜的组成部分和纤维素合酶活性等条目中。
此外,633个只在YM-2中表达的DEGs富集在DNA重组、转录调控、脂肪酸-氧化、逆转录转座子核衣壳和肽酶的活动等条目中,555个只在昌7-2中表达的DEGs富集在COPII囊泡包被的调节、核糖核苷二磷酸还原酶复合物和核酸酶活性等条目中。
2.5 YM-2和昌7-2中DEGs的KEGG分析
为进一步探究YM-2中调控亚油酸生物合成的途径,揭示其增强亚油酸生物合成的机制,对YM-2和昌7-2中的DEGs进行KEGG富集分析(图6)。具体来说,6505个DEGs被显著富集在亚油酸代谢、谷胱甘肽代谢、多种植物次生代谢产物的生物合成、黄酮类化合物的生物合成、二萜生物合成、甘油磷脂代谢芪类、二芳基庚烷和姜辣素生物合成、氰基氨基酸代谢、苯丙素生物合成、丙氨酸、天冬氨酸和谷氨酸代谢途径中。其中,LOC103642039、LOX1、LOX3、LOX6、LOX10、LOX12和TS1等7个基因被富集在亚油酸代谢途径中。
图6
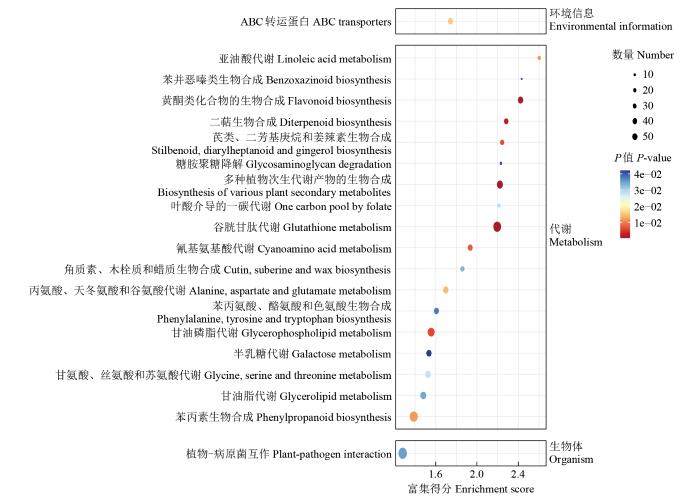
在YM-2中上调表达的3381个DEGs被显著富集在谷胱甘肽代谢、二萜生物合成、半胱氨酸和甲硫氨酸代谢、多种植物次生代谢产物的生物合成、芪类、二芳基庚烷和姜辣素生物合成、倍半萜和三萜生物合成、ABC转运蛋白、氰基氨基酸代谢、甘氨酸、丝氨酸和苏氨酸代谢以及丁酸代谢途径中。在YM-2中下调表达的3124个DEGs被显著富集在亚油酸代谢、黄酮类化合物的生物合成、多种植物次生代谢产物的生物合成、苯丙素生物合成、谷胱甘肽代谢、马达蛋白、甘油磷脂代谢、过氧化物酶体、糖胺聚糖降解和脂肪酸延长等途径中(图7)。其中,LOX1、LOX3、LOX6、LOX12和TS1等5个基因被富集在亚油酸代谢途径中。
图7
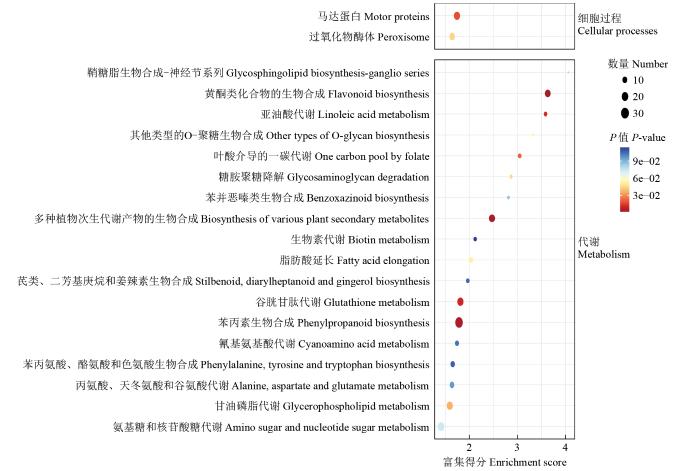
图7
3124个下调DEGs的KEGG富集分析
Fig.7
KEGG enrichment analysis of 3124 down-regulated DEGs
633个只在YM-2中表达的DEGs富集在苯丙氨酸、酪氨酸和色氨酸生物合成、脂肪酸的生物合成和柠檬酸循环(TCA循环)等条目中,其中,与脂肪酸的生物合成相关的LOC103646607只在YM-2中表达;555个只在昌7-2中表达的DEGs富集在苯丙素生物合成、谷胱甘肽代谢、花生四烯酸代谢以及氨基糖和核苷酸糖代谢等条目中。
综上所述,KEGG富集分析表明,YM-2与昌7-2的DEGs与亚油酸代谢和脂肪酸生物合成相关,候选基因LOX1、LOX3、LOX6、LOX10、LOX12、TS1、LOC103642039和LOC103646607可能与亚油酸的合成直接相关。
2.6 YM-2和昌7-2的亚油酸代谢通路分析
YM-2和昌7-2的DEGs被显著富集在亚油酸代谢途径(Zma00591)中。由图8可知,在亚油酸代谢途径中,YM-2中亚油酸9S-脂氧合酶(EC:1.13.11.58)下调表达,包括酶基因LOX1、LOX2、LOX3、LOX4、LOX5和LOX6,而亚油酸13S-脂氧合酶(EC:1.13.11.12)中下调表达的酶基因包括TS1和LOX12,上调表达的酶基因包括LOC103642039和LOX10,说明这些基因的差异表达与YM-2中亚油酸的含量密切相关。
图8
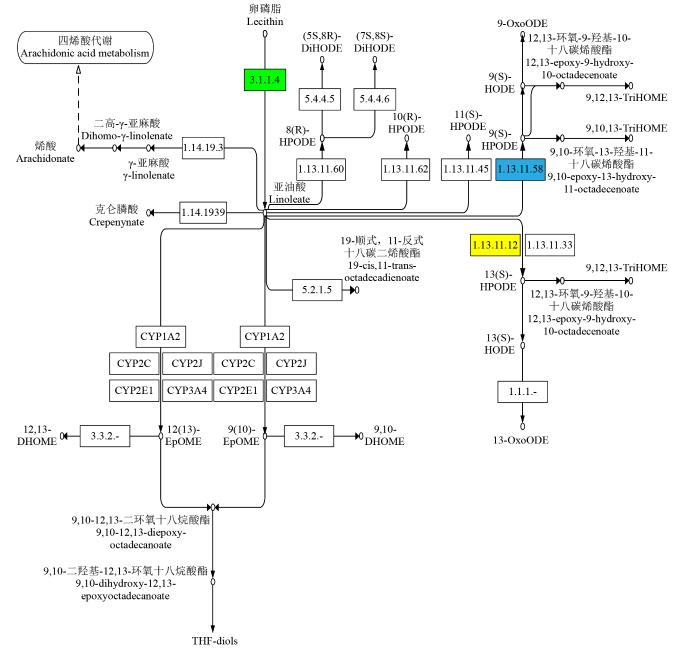
图8
亚油酸代谢KEGG通路
蓝色表示下调基因,黄色表示对应的基因中既有上调也有下调,绿色表示差异不显著。下同。
Fig.8
KEGG pathway of linoleic acid metabolism
Blue indicates down-regulated genes, yellow indicates that the corresponding gene contains both up-regulated and down-regulated expressions, and green indicates no significant difference. The same below.
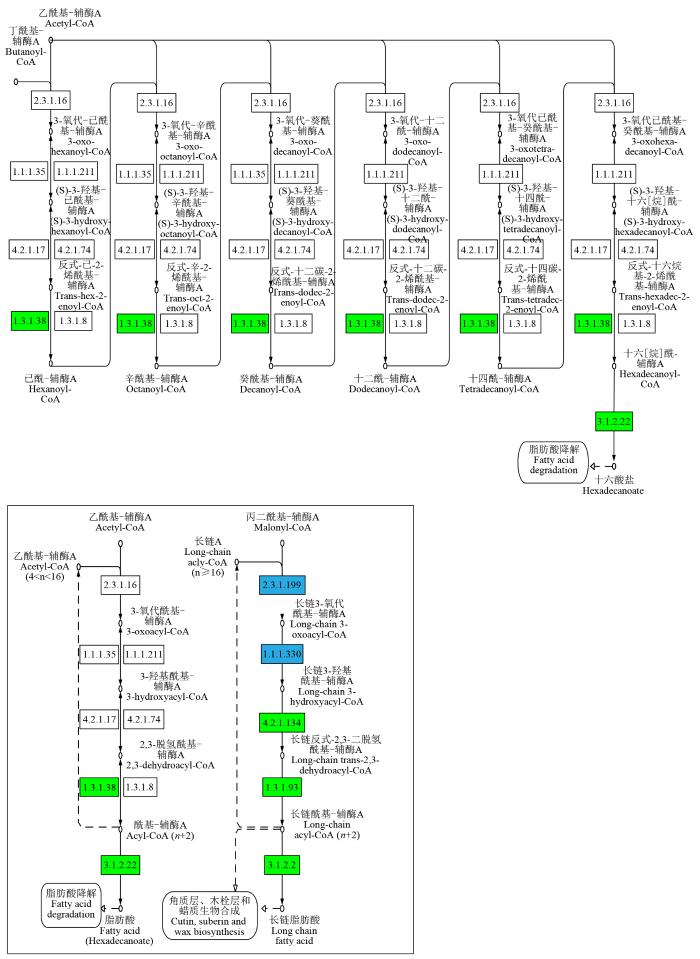
差异代谢物显著富集在脂肪酸延长途径(Zma00062)中,其中基因下调表达显著。由图9可知,3-酮脂酰-CoA合酶(EC:2.3.1.199)表达下调,包括酶基因LOC100272813、LOC100191738、LOC100285592、LOC542018和LOC103639429。同时,17β-雌二醇17-脱氢酶/极长链3-氧代酰基辅酶A还原酶(EC:1.1.1.62/1.1.1.330)表达下调,包括酶基因LOC103634953和LOC103653730。
图9
2.7 显著相反表达模式DEGs的qRT-PCR验证和功能分析
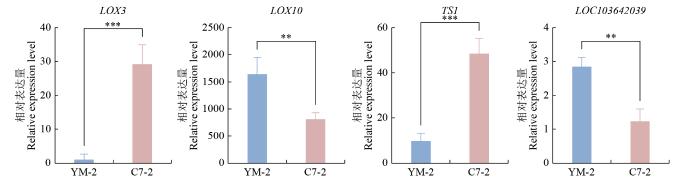
基于转录组测序分析,筛选出在YM-2与昌7-2中呈显著相反表达模式的4个脂氧合酶基因(lipoxygenase,LOX)进行qRT-PCR验证(表2)。这些基因均参与亚油酸代谢通路,且差异表达显著性通过多重检验校正(q<0.01)。根据转录组测序结果,自交系YM-2中LOX10和LOC103642039的基因表达量显著高于昌7-2,达昌7-2的2倍;而LOX3和TS1的表达量呈现相反趋势,在昌7-2中分别为YM-2的16倍和4倍,具有显著性差异。其中,LOX3和LOX10为脂加氧酶(LOX),是一组非血红素含铁双加氧酶,可催化多不饱和脂肪酸(PUFA)的氧化作用[20]。TS1编码一种具有质体靶向性的脂氧合酶,预测具有13-脂氧合酶特异性,LOX作为非血红素含铁脂肪酸双加氧酶,可催化亚油酸、α-亚麻酸和花生四烯酸等多不饱和脂肪酸的过氧化[21]。同时,参与脂质氧化的基因还包括LOC103642039,其编码脂氧合酶8。LOC103642039和LOX10在YM-2中表达量显著上调,LOX3和TS1则表现为下调,与转录组测序结果一致(图10)。值得注意的是,YM-2中LOX3基因表达量均显著低于昌7-2,可能在亚油酸生物合成中发挥重要的负调控作用。
表2 qRT-PCR选取的4个DEGs
Table 2
| 基因 Gene | 基因描述 Gene description | Log2FC | q值 q-value | 上调/下调 Up/down regulation | KEGG通路 KEGG pathway |
|---|---|---|---|---|---|
| LOX3 | 推测为亚油酸9S-脂氧合酶3 | -4.4152 | 3.50E-09 | 下调 | 亚油酸代谢 (Zma00591) |
| LOX10 | 脂氧合酶2.3,叶绿体 | 1.0281 | 9.57E-07 | 上调 | |
| TS1 | 推测为脂氧合酶5 | -2.2305 | 6.19E-24 | 下调 | |
| LOC103642039 | 可能为脂氧合酶8,叶绿体 | 1.2154 | 9.90E-06 | 上调 |
图10
3 讨论
3.1 YM-2和昌7-2中DEGs涉及的脂肪酸代谢途径
测序结果共检测到6505个DEGs,其中,3381个基因显著上调,3124个基因显著下调。从数据上看,本研究中构建了一个含有丰富差异代谢物信息的玉米转录组学数据库。YM-2中表达下调的3124个DEGs被显著富集在黄酮类化合物的生物合成、苯丙素生物合成、谷胱甘肽代谢、亚油酸代谢、甘油磷脂代谢、脂肪酸延长、氨基糖和核苷酸糖代谢、丙氨酸、天冬氨酸和谷氨酸代谢、氰基氨基酸代谢和苯丙氨酸、酪氨酸和色氨酸生物合成等途径中。633个只在YM-2中表达的DEGs富集在柠檬酸循环(TCA循环)、类胡萝卜素生物合成、氮代谢、苯丙氨酸、酪氨酸和色氨酸生物合成和脂肪酸的生物合成等条目中。研究[17]通过对超高油玉米品系与普通玉米品系的RNA-seq分析发现,DEGs与亚油酸代谢、氰基氨基酸代谢、谷胱甘肽代谢、丙氨酸、天冬氨酸代谢、谷氨酸代谢、氮代谢和类胡萝卜素途径显著相关。本研究进一步证实和补充了与玉米脂肪酸合成相关的重要途径,为油脂相关基因的挖掘提供参考。
KEGG富集结果发现,自主育成的自交系YM-2高产亚油酸表型明显。本研究重点关注了亚油酸代谢途径的关键基因,发现亚油酸9S-脂氧合酶(EC:1.13.11.58)和亚油酸13S-脂氧合酶(EC:1.13.11.12)基因显著下调。Xu等[22]通过蛋白质组学发现了13s-lipoxygenase-1、9s-lipoxygenase-1、9s-lipoxygenase-4和9s-lipoxygenase-5与亚油酸代谢密切相关,qRT-PCR分析发现其在亚油酸代谢中下调。结果表明本次转录组测序对基因表达量检测情况较为准确,可信度高,可作为理论参考。
3.2 YM-2中特异表达的DEGs
对YM-2和昌7-2样本之间的DEGs进行分析,发现633个DEGs只在YM-2中表达,555个DEGs只在昌7-2中表达,而5317个DEGs在2个样品中同时存在。研究重点关注在YM-2中特异表达的基因,GO富集分析表明其参与多种功能,包括转录调控、脂肪酸―氧化、韧皮部或木质部组织发生等途径。参与转录调控的基因包括LOC100304412、LOC100382585、LOC109943284、LOC111590845和LOC118472919,其中LOC109943284具有天冬氨酸型内肽酶活性。参与脂肪酸―氧化的基因包括LOC103629303和LOC109942761,均为过氧化物酶体(peroxisome),编码烯酰-CoA水合酶2。过氧化物酶体是与真核生物的生长、发育以及抗逆密切相关的多功能细胞器。在植物中,过氧化物酶体负责脂质动员、光呼吸、激素合成和活性氧清除等,对植物的发芽、光合、育性和抗性等生理过程至关重要[23]。参与韧皮部或木质部组织发生的基因包括LOC103637683、LOC109942137、LOC109943491和LOC118476038。LOC103637683为VAN3结合蛋白,参与生长素激活的信号通路、叶片维管组织模式形成和蔗糖的装载[24]。
在YM-2中特异表达的633个基因,经KEGG富集分析表明其与多种功能相关。其中,与氮代谢相关的基因LOC100384578编码α碳酸酐酶7,具有碳酸盐脱水酶活性和锌离子结合活性;基础转录因子LOC103630828编码转录起始因子TFIID亚基5,能够与蛋白质结合,使RNA聚合酶II具有一般转录起始因子的活性;苯丙氨酸、酪氨酸和色氨酸生物合成基因LOC103625743编码苯肽胺酸合成酶α亚基1,可激活邻氨基苯甲酸合酶活性并参与色氨酸生物合成过程。上述基因的挖掘可为其他功能研究提供参考。
3.3 候选基因可能参与玉米亚油酸合成和其他生物学途径
于NCBI数据库(
亚油酸酯9S-脂氧合酶3基因(LOX3)位于玉米1号染色体上,ORF长2595 bp,编码864个氨基酸,编码蛋白分子量大小为102.07 kDa。4种游离脂肪酸氧脂前体棕榈酸(C16:0)、油酸(C18:1)、亚油酸(C18:2)和亚麻酸(C18:3)在LOX3突变体中显著增加,而脂肪酸的积累也增加玉米对曲霉菌属的敏感性[27]。
4 结论
本研究利用转录组学,从自主育成的高亚油酸玉米自交系YM-2和普通玉米自交系昌7-2的苗期叶片中共检测到6505个DEGs,其中633个DEGs只在YM-2中表达。DEGs被显著富集在亚油酸代谢、谷胱甘肽代谢、多种植物次生代谢产物的生物合成、黄酮类化合物的生物合成、二萜生物合成、甘油磷脂代谢芪类、二芳基庚烷和姜辣素生物合成、氰基氨基酸代谢、苯丙素生物合成和丙氨酸、天冬氨酸和谷氨酸代谢途径中。其中,8个基因LOX1、LOX3、LOX6、LOX10、LOX12、TS1、LOC103642039和LOC103646607被富集在亚油酸代谢途径和脂肪酸的生物合成中。
参考文献
大胚高油玉米籽粒蛋白质含量相关QTLs定位研究
Brain docosahexaenoic acid uptake and metabolism
DOI:S0098-2997(17)30135-8
PMID:29305120
[本文引用: 1]

Docosahexaenoic acid (DHA) is the most abundant n-3 polyunsaturated fatty acid in the brain where it serves to regulate several important processes and, in addition, serves as a precursor to bioactive mediators. Given that the capacity of the brain to synthesize DHA locally is appreciably low, the uptake of DHA from circulating lipid pools is essential to maintaining homeostatic levels. Although, several plasma pools have been proposed to supply the brain with DHA, recent evidence suggests non-esterified-DHA and lysophosphatidylcholine-DHA are the primary sources. The uptake of DHA into the brain appears to be regulated by a number of complementary pathways associated with the activation and metabolism of DHA, and may provide mechanisms for enrichment of DHA within the brain. Following entry into the brain, DHA is esterified into and recycled amongst membrane phospholipids contributing the distribution of DHA in brain phospholipids. During neurotransmission and following brain injury, DHA is released from membrane phospholipids and converted to bioactive mediators which regulate signaling pathways important to synaptogenesis, cell survival, and neuroinflammation, and may be relevant to treating neurological diseases. In the present review, we provide a comprehensive overview of brain DHA metabolism, encompassing many of the pathways and key enzymatic regulators governing brain DHA uptake and metabolism. In addition, we focus on the release of non-esterified DHA and subsequent production of bioactive mediators and the evidence of their proposed activity within the brain. We also provide a brief review of the evidence from post-mortem brain analyses investigating DHA levels in the context of neurological disease and mood disorder, highlighting the current disparities within the field.Copyright © 2018 The Authors. Published by Elsevier Ltd.. All rights reserved.
Genome-wide association study dissects the genetic architecture of oil biosynthesis in maize kernels
DOI:10.1038/ng.2484
PMID:23242369
[本文引用: 1]
Maize kernel oil is a valuable source of nutrition. Here we extensively examine the genetic architecture of maize oil biosynthesis in a genome-wide association study using 1.03 million SNPs characterized in 368 maize inbred lines, including 'high-oil' lines. We identified 74 loci significantly associated with kernel oil concentration and fatty acid composition (P < 1.8 × 10(-6)), which we subsequently examined using expression quantitative trait loci (QTL) mapping, linkage mapping and coexpression analysis. More than half of the identified loci localized in mapped QTL intervals, and one-third of the candidate genes were annotated as enzymes in the oil metabolic pathway. The 26 loci associated with oil concentration could explain up to 83% of the phenotypic variation using a simple additive model. Our results provide insights into the genetic basis of oil biosynthesis in maize kernels and may facilitate marker-based breeding for oil quantity and quality.
Expression of ZmLEC1 and ZmWRI1 increases seed oil production in maize
DOI:10.1104/pp.110.157537
PMID:20488892
[本文引用: 1]
Increasing seed oil production is a major goal for global agriculture to meet the strong demand for oil consumption by humans and for biodiesel production. Previous studies to increase oil synthesis in plants have focused mainly on manipulation of oil pathway genes. As an alternative to single-enzyme approaches, transcription factors provide an attractive solution for altering complex traits, with the caveat that transcription factors may face the challenge of undesirable pleiotropic effects. Here, we report that overexpression of maize (Zea mays) LEAFY COTYLEDON1 (ZmLEC1) increases seed oil by as much as 48% but reduces seed germination and leaf growth in maize. To uncouple oil increase from the undesirable agronomic traits, we identified a LEC1 downstream transcription factor, maize WRINKLED1 (ZmWRI1). Overexpression of ZmWRI1 results in an oil increase similar to overexpression of ZmLEC1 without affecting germination, seedling growth, or grain yield. These results emphasize the importance of field testing for developing a commercial high-oil product and highlight ZmWRI1 as a promising target for increasing oil production in crops.
Genome-wide association analysis-based mining of quality genes related to linoleic and linolenic acids in soybean
Genome-wide association study identifies candidate genes related to the linoleic acid content in soybean seeds
Overexpression of miR319a altered oil body morphogenesis and lipid content in Arabidopsis seeds
Single-cell transcriptomes reveal spatiotemporal heat stress response in maize roots
Utilizing transcriptomics and metabolomics to unravel key genes and metabolites of maize seedlings in response to drought stress
DOI:10.1186/s12870-023-04712-y
PMID:38185653
Drought stress can substantially restrict maize growth and productivity, and global warming and an increasing frequency of extreme weather events are likely to result in more yield losses in the future. Therefore, unraveling the molecular mechanism underlying the response to drought stress is essential for breeding drought-resilient crops.In this study, we subjected the 3-leaf-period plants of two maize inbred lines, a drought-tolerant line (si287) and a drought-sensitive line (X178), to drought stress for seven days while growing in a chamber. Subsequently, we measured physiological traits and analyzed transcriptomic and metabolic profiles of two inbred lines. Our KEGG analysis of genes and metabolites revealed significant differences in pathways related to glycolysis/gluconeogenesis, flavonoid biosynthesis, starch and sucrose metabolism, and biosynthesis of amino acids. Additionally, our joint analysis identified proline, tryptophan and phenylalanine are crucial amino acids for maize response to drought stress. Furthermore, we concentrated on tryptophan (Trp), which was found to enhance tolerance via IAA-ABA signaling, as well as SA and nicotinamide adenine dinucleotide (NAD) consequent reactive oxygen species (ROS) scavenging. We identified three hub genes in tryptophan biosynthesis, indole-3-acetaldehyde oxidase (ZmAO1, 542,228), catalase 1 (ZmCAT1, 542,369), and flavin-containing monooxygenase 6 (ZmYUC6, 103,629,142), High expression of these genes plays a significant role in regulating drought tolerance. Two metabolites related to tryptophan biosynthesis, quinolinic acid, and kynurenine improved maize tolerance to drought stress by scavenging reactive oxygen species.This study illuminates the mechanisms underlying the response of maize seedlings to drought stress. Especially, it identifies novel candidate genes and metabolites, enriching our understanding of the role of tryptophan in drought stress. The identification of distinct resistance mechanisms in maize inbred lines will facilitate the exploration of maize germplasm and the breeding of drought-resilient hybrids.© 2024. The Author(s).
Gene expression and expression quantitative trait loci analyses uncover natural variations underlying the improvement of important agronomic traits during modern maize breeding
DOI:10.1111/tpj.16260
PMID:37186341
Maize (Zea mays L.) is a major staple crop worldwide, and during modern maize breeding, cultivars with increased tolerance to high-density planting and higher yield per plant have contributed significantly to the increased yield per unit land area. Systematically identifying key agronomic traits and their associated genomic changes during modern maize breeding remains a significant challenge because of the complexity of genetic regulation and the interactions of the various agronomic traits, with most of them being controlled by numerous small-effect quantitative trait loci (QTLs). Here, we performed phenotypic and gene expression analyses for a set of 137 elite inbred lines of maize from different breeding eras in China. We found four yield-related traits are significantly improved during modern maize breeding. Through gene-clustering analyses, we identified four groups of expressed genes with distinct trends of expression pattern change across the historical breeding eras. In combination with weighted gene co-expression network analysis, we identified several candidate genes regulating various plant architecture- and yield-related agronomic traits, such as ZmARF16, ZmARF34, ZmTCP40, ZmPIN7, ZmPYL10, ZmJMJ10, ZmARF1, ZmSWEET15b, ZmGLN6 and Zm00001d019150. Further, by combining expression quantitative trait loci (eQTLs) analyses, correlation coefficient analyses and population genetics, we identified a set of candidate genes that might have been under selection and contributed to the genetic improvement of various agronomic traits during modern maize breeding, including a number of known key regulators of plant architecture, flowering time and yield-related traits, such as ZmPIF3.3, ZAG1, ZFL2 and ZmBES1. Lastly, we validated the functional variations in GL15, ZmPHYB2 and ZmPYL10 that influence kernel row number, flowering time, plant height and ear height, respectively. Our results demonstrates the effectiveness of our combined approaches for uncovering key candidate regulatory genes and functional variation underlying the improvement of important agronomic traits during modern maize breeding, and provide a valuable genetic resource for the molecular breeding of maize cultivars with tolerance for high-density planting.© 2023 Society for Experimental Biology and John Wiley & Sons Ltd.
Genetic basis of the oil biosynthesis in ultra-high-oil maize grains with an oil content exceeding 20%
Duplicate maize Wrinkled1 transcription factors activate target genes involved in seed oil biosynthesis
KEGG for linking genomes to life and the environment
Characterization of the maize lipoxygenase gene family in relation to aflatoxin accumulation resistance
tasselseed1 is a lipoxygenase affecting jasmonic acid signaling in sex determination of maize
DOI:10.1126/science.1164645
PMID:19131630
[本文引用: 3]
Sex determination in maize is controlled by a developmental cascade leading to the formation of unisexual florets derived from an initially bisexual floral meristem. Abortion of pistil primordia in staminate florets is controlled by a tasselseed-mediated cell death process. We positionally cloned and characterized the function of the sex determination gene tasselseed1 (ts1). The TS1 protein encodes a plastid-targeted lipoxygenase with predicted 13-lipoxygenase specificity, which suggests that TS1 may be involved in the biosynthesis of the plant hormone jasmonic acid. In the absence of a functional ts1 gene, lipoxygenase activity was missing and endogenous jasmonic acid concentrations were reduced in developing inflorescences. Application of jasmonic acid to developing inflorescences rescued stamen development in mutant ts1 and ts2 inflorescences, revealing a role for jasmonic acid in male flower development in maize.
Quantitative proteomic and lipidomics analyses of high oil contentGmDGAT1-2 transgenic soybean illustrate the regulatory mechanism of lipoxygenase and oleosin
Peroxisomes: versatile organelles with diverse roles in plants
DOI:10.1111/nph.16134
PMID:31442305
[本文引用: 1]
Peroxisomes are small, ubiquitous organelles that are delimited by a single membrane and lack genetic material. However, these simple-structured organelles are highly versatile in morphology, abundance and protein content in response to various developmental and environmental cues. In plants, peroxisomes are essential for growth and development and perform diverse metabolic functions, many of which are carried out coordinately by peroxisomes and other organelles physically interacting with peroxisomes. Recent studies have added greatly to our knowledge of peroxisomes, addressing areas such as the diverse proteome, regulation of division and protein import, pexophagy, matrix protein degradation, solute transport, signaling, redox homeostasis and various metabolic and physiological functions. This review summarizes our current understanding of plant peroxisomes, focusing on recent discoveries. Current problems and future efforts required to better understand these organelles are also discussed. An improved understanding of peroxisomes will be important not only to the understanding of eukaryotic cell biology and metabolism, but also to agricultural efforts aimed at improving crop performance and defense.© 2019 The Authors. New Phytologist © 2019 New Phytologist Trust.
Phloem loading and unloading of sucrose: what a long, strange trip from source to sink
DOI:10.1146/annurev-arplant-070721-083240
PMID:35171647
[本文引用: 1]
Sucrose is transported from sources (mature leaves) to sinks (importing tissues such as roots, stems, fruits, and seeds) through the phloem tissues in veins. In many herbaceous crop species, sucrose must first be effluxed to the cell wall by a sugar transporter of the SWEET family prior to being taken up into phloem companion cells or sieve elements by a different sugar transporter, called SUT or SUC. The import of sucrose into these cells is termed apoplasmic phloem loading. In sinks, sucrose can similarly exit the phloem apoplasmically or, alternatively, symplasmically through plasmodesmata into connecting parenchyma storage cells. Recent advances describing the regulation and manipulation of sugar transporter expression and activities provide stimulating new insights into sucrose phloem loading in sources and unloading processes in sink tissues. Additionally, new breakthroughs have revealed distinct subpopulations of cells in leaves with different functions pertaining to phloem loading. These and other discoveries in sucrose transport are discussed. Expected final online publication date for the, Volume 73 is May 2022. Please see http://www.annualreviews.org/page/journal/pubdates for revised estimates.
The lipoxygenase pathway
Lipid peroxidation is common to all biological systems, both appearing in developmentally and environmentally regulated processes of plants. The hydroperoxy polyunsaturated fatty acids, synthesized by the action of various highly specialized forms of lipoxygenases, are substrates of at least seven different enzyme families. Signaling compounds such as jasmonates, antimicrobial and antifungal compounds such as leaf aldehydes or divinyl ethers, and a plant-specific blend of volatiles including leaf alcohols are among the numerous products. Cloning of many lipoxygenases and other key enzymes within the lipoxygenase pathway, as well as analyses by reverse genetic and metabolic profiling, revealed new reactions and the first hints of enzyme mechanisms, multiple functions, and regulation. These aspects are reviewed with respect to activation of this pathway as an initial step in the interaction of plants with pathogens, insects, or abiotic stress and at distinct stages of development.
Maize 9-lipoxygenase ZmLOX3 controls development, root-specific expression of defense genes, and resistance to root-knot nematodes
Inactivation of the lipoxygenase ZmLOX3 increases susceptibility of maize to Aspergillus spp.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}