


胚胎发育晚期丰富蛋白(late embryogenesis abundant protein,LEA)是植物种子发育后期积累的小分子特异性肽,与种子寿命、胚乳质量和干燥耐受性相关[1-2],其作为种子成熟的标志之一,最早发现于棉花(Gossypium herbaceum L.)子叶中[3]。LEA蛋白富含甘氨酸,缺少色氨酸,具有高度亲水性,在减少水分流失和建立脱水耐受机制方面发挥关键作用[4];其兼具极强的耐热性和耐酸性,参与应对逆境的相关生理过程,有助于提升植物在各类生境压力下的耐受性[5-6]。此外,LEA蛋白可以通过结合二价阳离子重新定向水分,有效抑制过氧化氢酶、乳酸脱氢酶和苹果酸脱氢酶的活性,从而提升细胞的活性氧清除能力[7-8]。LEA蛋白存在于叶、茎和根等植物组织中[5],受多种非生物胁迫诱导,参与植物逆境应答机制[9]。Yang等[10]发现,小麦TaLEA2-1基因在根部特异性表达,其过表达可以显著提升根的长度和粗度,增加过氧化氢酶的活性,提升转基因小麦对盐胁迫的耐受性。Cheng等[11]在杨树(Populus trichocarpa)中发现了88个LEA基因,于根、茎和叶中分别检测到24、22和19个LEA基因受盐胁迫诱导表达,表明LEA基因在杨树生长发育及盐胁迫响应过程中发挥重要功能。
藜麦(Chenopodium quinoa Willd.)是藜属(Chenopodium Linn.)异源四倍体一年生草本植物,具有较强的生境适应能力,能够在贫瘠土壤中保持农业生产力,特别是在高盐(电导率40 dS/m)环境下仍生长良好[12-13]。随着藜麦全基因组数据[14]的公布,利用生物信息学手段在全基因组水平上鉴定和分析LEA基因家族成为可能。本研究基于Pfam数据库[15]中LEA家族7个亚组(LEA_1、LEA_2、LEA_3a、LEA_3b、LEA_4、LEA_5、LEA_6)的HMM模型,在藜麦基因组水平进行鉴定,对成员间的系统发育、基因结构、染色体分布、串联重复基因和转录表达等信息进行系统分析,为进一步利用该家族进行藜麦抗逆性改良提供参考。
1 材料与方法
1.1 试验材料
参试材料为青藜4号(青认备2019001)。
1.2 试验方法
1.2.1 藜麦LEA基因家族成员鉴定
在Quinoa Genome数据库(
1.2.2 藜麦LEA蛋白的理化性质分析
利用ExPASy ProtParam(
1.2.3 藜麦LEA蛋白家族成员系统发育树构建
利用MEGA 7.0中邻接(Neighbor-joining)法,设置Bootstrap值为1000次,对HMMER(
1.2.4 藜麦LEA基因家族成员保守结构域预测及基因结构分析
使用GSDS 2.0(
1.2.5 藜麦LEA基因家族成员染色体定位及共线性分析
利用在线软件MG2C(
1.2.6 转录组测序及qRT-PCR
选取籽粒饱满、大小一致的藜麦种子,采用75%的乙醇消毒5 min后,无菌水冲洗至干净无味,用吸水纸吸取种子表面水分,平铺于放有2层滤纸的种子萌发盒中,加入15 mL ddH2O,置于12 h(25 ℃)光照、12 h(20 ℃)黑暗条件的人工气候培养箱中萌发。1 d后,选取生长状况良好且一致的幼苗进行300 mmol/L NaCl胁迫处理,分别于0、24、48和72 h整株取样,液氮中迅速冷冻,-80 ℃保存备用,委托生工生物工程(上海)股份有限公司进行转录组测序。转录本数据上传NCBI数据库(
表1 藜麦LEA基因实时荧光定量引物
Table 1
| 基因ID Gene ID | 上游引物Forward primer (5'-3') | 下游引物Reverse primer (5'-3') |
|---|---|---|
| AUR62012039 | AGGAAAGAAAGCAGTAGAGTCC | AGTGGTGGAGTGAGTGTGAG |
| AUR62040787 | ACCGATGAACCCGAGAATC | TTAGCCTGTCAACCGAGTAGC |
| AUR62008681 | CGCTCTCTATCCAACGCTAAG | TCAGCAACATCAATCTCAGC |
| AUR62025562 | TGGCTCGCTCTCTATTCAAC | CCTGTAGTATCCAGTTACGGG |
| AUR62018884 | CAAATCGTTTCTCACCCAAC | AAAGGTCTTCTGCTCCAGC |
| AUR62005994 | CATTTGGTTCATCCTCCAC | TGTTGCCCTTTGTAAGAAGC |
| LOC110715281 (ACT-1)[16] | GTCCACAGAAAGTGCTTCTAAG | AACAACTCCTCACCTTCTCATG |
1.2.7 藜麦LEA基因家族成员启动子分析
在藜麦基因组数据库中截取LEA基因起始密码子上游1500 bp序列,利用植物顺势作用元件数据库PlantCare(
1.3 数据处理
采用Excel 2010进行统计分析,GraphPad 9.2作图。
2 结果与分析
2.1 藜麦LEA蛋白理化性质分析及亚细胞定位预测
图1
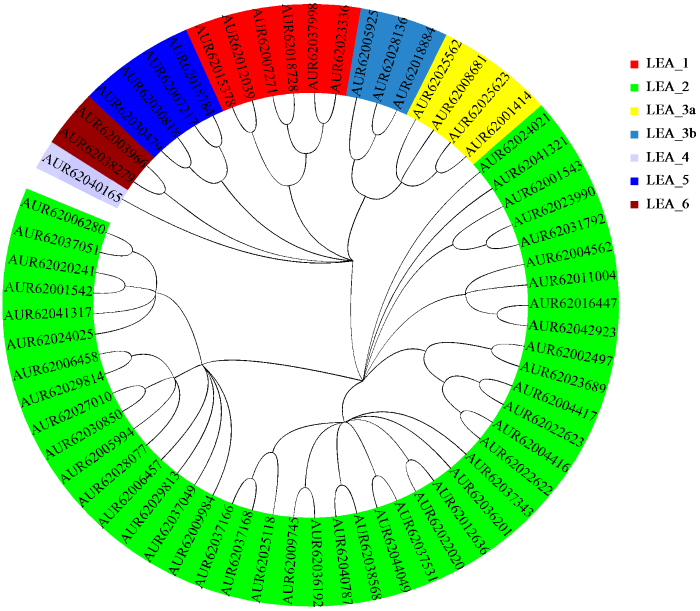
表2 藜麦LEA蛋白的理化性质及亚细胞定位
Table 2
| 蛋白ID Protein ID | 氨基酸长度 Amino acid length | 分子量 Molecular weight (Da) | 等电点 pI | 亲水性指数 Hydropathy index | 亚细胞定位预测 Subcellular localization prediction | 亚组 Subgroup | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AUR62015378 | 585 | 62 135.91 | 6.16 | -0.905 | 细胞核 | LEA_1 | ||||||
| AUR62012039 | 185 | 18 965.61 | 9.16 | -0.941 | 细胞核 | LEA_1 | ||||||
| AUR62007271 | 135 | 14 669.53 | 9.46 | -0.916 | 细胞核 | LEA_1 | ||||||
| AUR62018728 | 133 | 14 474.30 | 9.52 | -0.932 | 细胞核 | LEA_1 | ||||||
| AUR62037998 | 411 | 46 980.05 | 9.10 | -0.148 | 细胞核 | LEA_1 | ||||||
| AUR62023336 | 66 | 7632.58 | 5.94 | -1.315 | 细胞核 | LEA_1 | ||||||
| AUR62024021 | 195 | 22 461.13 | 9.73 | -0.282 | 叶绿体 | LEA_2 | ||||||
| AUR62041321 | 195 | 22 560.25 | 9.67 | -0.335 | 叶绿体 | LEA_2 | ||||||
| AUR62001543 | 265 | 30 001.52 | 9.49 | 0.010 | 细胞膜 | LEA_2 | ||||||
| AUR62023990 | 235 | 26 515.15 | 9.78 | -0.234 | 细胞膜 | LEA_2 | ||||||
| AUR62031792 | 235 | 26 387.11 | 9.87 | -0.208 | 细胞膜 | LEA_2 | ||||||
| AUR62004562 | 253 | 27 621.05 | 10.56 | -0.245 | 细胞核 | LEA_2 | ||||||
| AUR62011004 | 134 | 14 815.91 | 5.26 | 0.050 | 叶绿体 | LEA_2 | ||||||
| AUR62016447 | 221 | 25 098.34 | 9.55 | -0.133 | 叶绿体 | LEA_2 | ||||||
| AUR62042923 | 221 | 25 233.27 | 9.32 | -0.138 | 叶绿体 | LEA_2 | ||||||
| AUR62002497 | 385 | 42 855.48 | 5.99 | -0.321 | 叶绿体 | LEA_2 | ||||||
| AUR62023689 | 369 | 41 236.24 | 5.83 | -0.459 | 细胞质 | LEA_2 | ||||||
| AUR62004417 | 327 | 36 389.79 | 6.88 | -0.233 | 细胞核 | LEA_2 | ||||||
| AUR62022623 | 261 | 29 129.75 | 8.81 | -0.141 | 叶绿体 | LEA_2 | ||||||
| AUR62004416 | 159 | 17 883.58 | 5.94 | -0.311 | 叶绿体 | LEA_2 | ||||||
| AUR62022622 | 159 | 17 911.63 | 5.94 | -0.313 | 叶绿体 | LEA_2 | ||||||
| AUR62037343 | 230 | 26 081.43 | 8.38 | 0.061 | 叶绿体 | LEA_2 | ||||||
| AUR62036201 | 397 | 42 223.68 | 6.90 | -0.251 | 细胞质 | LEA_2 | ||||||
| AUR62012636 | 625 | 70 739.43 | 7.36 | -0.342 | 叶绿体 | LEA_2 | ||||||
| AUR62022020 | 907 | 100 918.35 | 9.26 | -0.264 | 叶绿体 | LEA_2 | ||||||
| AUR62037531 | 257 | 28 396.07 | 9.68 | -0.202 | 细胞核 | LEA_2 | ||||||
| AUR62044049 | 260 | 28 782.62 | 9.84 | -0.165 | 线粒体 | LEA_2 | ||||||
| AUR62038568 | 260 | 28 969.89 | 9.95 | -0.105 | 细胞核 | LEA_2 | ||||||
| AUR62040787 | 260 | 29 063.15 | 9.89 | -0.073 | 细胞膜 | LEA_2 | ||||||
| AUR62036192 | 201 | 22 519.08 | 9.37 | -0.090 | 细胞核 | LEA_2 | ||||||
| AUR62009745 | 202 | 22 541.10 | 9.37 | -0.084 | 叶绿体 | LEA_2 | ||||||
| AUR62025118 | 411 | 44 859.42 | 9.83 | 0.009 | 细胞膜 | LEA_2 | ||||||
| AUR62037168 | 294 | 32 124.13 | 9.74 | -0.330 | 叶绿体 | LEA_2 | ||||||
| AUR62037166 | 294 | 32 124.13 | 9.74 | -0.330 | 叶绿体 | LEA_2 | ||||||
| AUR62009984 | 362 | 40 962.60 | 9.28 | -0.409 | 叶绿体 | LEA_2 | ||||||
| AUR62037049 | 235 | 26 528.57 | 9.15 | -0.274 | 叶绿体 | LEA_2 | ||||||
| AUR62029813 | 193 | 20 922.48 | 10.08 | 0.216 | 叶绿体 | LEA_2 | ||||||
| AUR62006457 | 193 | 20 962.50 | 10.02 | 0.210 | 细胞核 | LEA_2 | ||||||
| AUR62028077 | 506 | 55 181.12 | 5.71 | -0.133 | 细胞壁 | LEA_2 | ||||||
| AUR62005994 | 210 | 23 568.40 | 9.40 | -0.006 | 细胞膜 | LEA_2 | ||||||
| AUR62030850 | 727 | 83 308.07 | 6.24 | -0.230 | 叶绿体 | LEA_2 | ||||||
| AUR62027010 | 209 | 22 664.20 | 9.32 | 0.171 | 细胞膜 | LEA_2 | ||||||
| AUR62029814 | 211 | 23 171.20 | 9.97 | 0.120 | 叶绿体 | LEA_2 | ||||||
| AUR62006458 | 211 | 23 160.13 | 9.87 | 0.096 | 叶绿体 | LEA_2 | ||||||
| AUR62024025 | 169 | 19 641.29 | 7.79 | -0.349 | 过氧化物酶体 | LEA_2 | ||||||
| AUR62041317 | 217 | 24 772.49 | 9.14 | -0.156 | 细胞壁 | LEA_2 | ||||||
| AUR62001542 | 212 | 23 801.43 | 9.49 | -0.028 | 叶绿体 | LEA_2 | ||||||
| AUR62020241 | 214 | 24 051.61 | 9.54 | -0.009 | 叶绿体 | LEA_2 | ||||||
| AUR62037051 | 209 | 23 402.50 | 9.68 | 0.176 | 细胞膜 | LEA_2 | ||||||
| AUR62006280 | 256 | 29 111.35 | 10.44 | -0.128 | 叶绿体 | LEA_2 | ||||||
| AUR62025562 | 93 | 10 039.37 | 9.89 | -0.357 | 叶绿体 | LEA_3a | ||||||
| AUR62008681 | 93 | 9969.23 | 9.89 | -0.387 | 叶绿体 | LEA_3a | ||||||
| AUR62025623 | 101 | 11 655.31 | 9.69 | -0.517 | 叶绿体 | LEA_3a | ||||||
| AUR62001414 | 100 | 11 431.16 | 9.77 | -0.380 | 叶绿体 | LEA_3a | ||||||
| AUR62005925 | 125 | 13 672.20 | 7.91 | -0.582 | 细胞核 | LEA_3b | ||||||
| AUR62028136 | 164 | 17 893.23 | 6.74 | -0.235 | 细胞核 | LEA_3b | ||||||
| AUR62018884 | 125 | 13 633.10 | 6.96 | -0.648 | 细胞核 | LEA_3b | ||||||
| AUR62040165 | 498 | 53 522.13 | 6.87 | -1.016 | 细胞核 | LEA_4 | ||||||
| AUR62030479 | 114 | 12 430.48 | 5.77 | -1.383 | 细胞核 | LEA_5 | ||||||
| AUR62030818 | 114 | 12 433.50 | 6.16 | -1.403 | 细胞核 | LEA_5 | ||||||
| AUR62007217 | 84 | 9075.84 | 5.92 | -1.585 | 细胞核 | LEA_5 | ||||||
| AUR62018784 | 84 | 8950.71 | 5.90 | -1.474 | 细胞核 | LEA_5 | ||||||
| AUR62038279 | 87 | 9085.71 | 4.46 | -1.171 | 细胞核 | LEA_6 | ||||||
| AUR62003966 | 87 | 9126.81 | 4.59 | -1.183 | 细胞核 | LEA_6 | ||||||
LEA_2亚组包含44个蛋白,长度为134~907个氨基酸,分子量为14 815.91~100 918.35 Da,等电点为5.26~10.56;亲水性指数为-0.459~0.216,仅有10个为疏水蛋白,亚细胞定位预测50%以上位于叶绿体,少量分布于细胞膜、细胞核、细胞壁和线粒体中。LEA_3a亚组包含4个蛋白,长度为93~101个氨基酸,分子量为9969.23~11 655.31 Da,等电点为9.69~9.89;亲水性指数为-0.517~-0.357,均为亲水性蛋白,亚细胞定位预测均位于叶绿体。LEA_3b亚组包含3个蛋白,长度为125~164个氨基酸,分子量为13 633.10~17 893.23 Da,等电点为6.74~7.91;亲水性指数为-0.648~-0.235,均为亲水性蛋白,亚细胞定位预测均位于细胞核。LEA_4亚组仅包含1个蛋白,长度为498个氨基酸,分子量为53 522.13 Da,等电点为6.87;亲水性指数为-1.016,为亲水蛋白,亚细胞定位预测位于细胞核。LEA_5亚组包含4个蛋白,长度为84~114个氨基酸,分子量为8950.71~12 433.50 Da,等电点为5.77~6.16;亲水性指数为-1.585~-1.383,均为亲水性蛋白,亚细胞定位预测均位于细胞核。LEA_6亚组包含2个蛋白,长度均为87个氨基酸,分子量分别为9085.71和9126.81 Da,等电点分别为4.46和4.59;亲水性指数分别为-1.171和-1.183,均为亲水性蛋白,亚细胞定位预测均位于细胞核。
2.2 藜麦LEA基因家族成员的基因结构及蛋白motif分析
根据LEA基因的核酸和蛋白质序列信息分别绘制基因结构和预测保守结构域(图2)。结果显示,LEA_1亚组包含0~2个内含子,仅有AUR62037998蛋白含有motif 6。LEA_2亚组包含0~8个内含子,蛋白序列N端多以motif 1、5、7和10为主,C端多以motif 3和8为主。LEA_3a和LEA_3b亚组包含1~2个内含子,蛋白序列仅包含motif 9。LEA_4亚组包含2个内含子,蛋白序列并未发现motif存在。LEA_5亚组均包含1个内含子,蛋白序列均只含有motif 6。LEA_6亚组不存在内含子,蛋白序列也未发现motif存在。总体而言,同一亚组中LEA基因的外显子―内含子和结构相似;不同亚组编码的蛋白中motif种类和数量存在较大的差异,但是同一亚组中的蛋白具有相似的motif结构。
图2
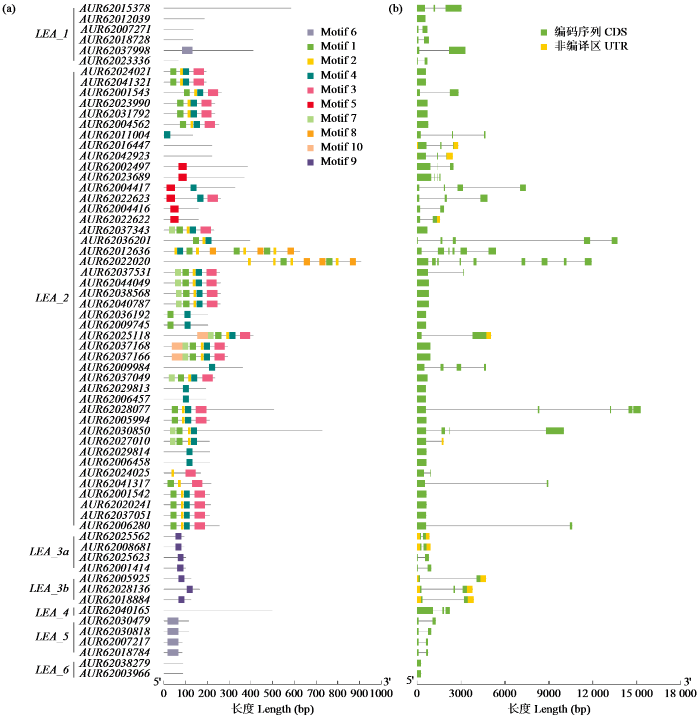
图2
藜麦LEA基因结构及motif分析
Fig.2
Gene structure and motif analysis of LEA genes in quinoa
2.3 藜麦LEA基因的染色体分布及基因复制分析
根据公共数据库公布的注释信息,绘制每个LEA基因在所属染色体的位置(图3),分析藜麦LEA家族的遗传分化和基因复制。藜麦共包含18条染色体,除了03、04、05、06和14号染色体以外,其他13条染色体上分布有62个LEA基因。LEA基因在染色体上的分布不均匀,其中07号染色体上有14个LEA基因,而08、09和12号染色体都仅有1个LEA基因。此外,并未发现单独亚组成簇分布的现象,表明LEA基因的复制和进化在染色体层面具有跳跃性。由于藜麦基因组序列不完整,AUR62038279和AUR62044049分布在未知染色体上,但对结果未产生较大影响。
图3

图3
藜麦LEA基因的染色体定位分析
Fig.3
Genomic distribution of LEA genes on quinoa chromosomes
在藜麦基因组上共发现11对同源基因和9对串联重复基因(图4)。其中LEA_1、LEA_3b和LEA_6亚组均仅有1对同源基因,LEA_3a和LEA_5亚组各有2对同源基因,LEA_2亚组含有5对同源基因和9对串联重复基因。对所有同源基因对和串联重复基因对进行Ka/Ks计算,结果均小于1,表明LEA基因在进化过程中可能经历了净化选择压力,也说明重复基因的结构稳定且进化保守。
图4

图4
藜麦LEA基因家族成员共线性分析
内圈数字表示染色体序号。
Fig.4
Collinear distribution of LEA gene family members in quinoa
Numbers in inner circle indicate chromosome number.
2.4 藜麦LEA基因物种间共线性分析
分别对藜麦与拟南芥、水稻、蒺藜苜蓿和大豆进行LEA基因同源性分析(图5)。藜麦与蒺藜苜蓿和大豆分别鉴定出49和95对LEA同源基因,与拟南芥和水稻分别鉴定出32和7对LEA同源基因。藜麦与豆科植物之间的LEA同源基因对数量较多,十字花科次之,禾本科最少。表明藜麦与豆科植物之间LEA基因具有高度的同源性。
图5

图5
藜麦与拟南芥、水稻、蒺藜苜蓿和大豆中LEA基因共线性分析
红线为LEA共线性基因对,灰线为其他基因的共线性。数字表示上述作物基因组的染色体数量。
Fig.5
Collinear analysis of LEA genes in quinoa with Arabidopsis thaliana, Oryza sativa, Medicago truncatula and Glycine max
The red line is the collinearity of LEAs, and the gray line is the collinearity of other genes. The numerals represent the chromosome numbers of the genomes of the above crops.
2.5 藜麦LEA基因家族成员启动子顺式作用元件分析
通过对LEA基因上游1500 bp启动子区域进行顺式作用元件预测,发现64个LEA基因的启动子顺式作用元件均与激素(脱落酸、生长素、赤霉素、水杨酸、茉莉酸甲酯)、干旱、低温、厌氧和防御有关(图6)。表明LEA基因家族成员通过不同的途径参与各种非生物胁迫应答,在藜麦的生长发育过程中起到关键作用。
图6
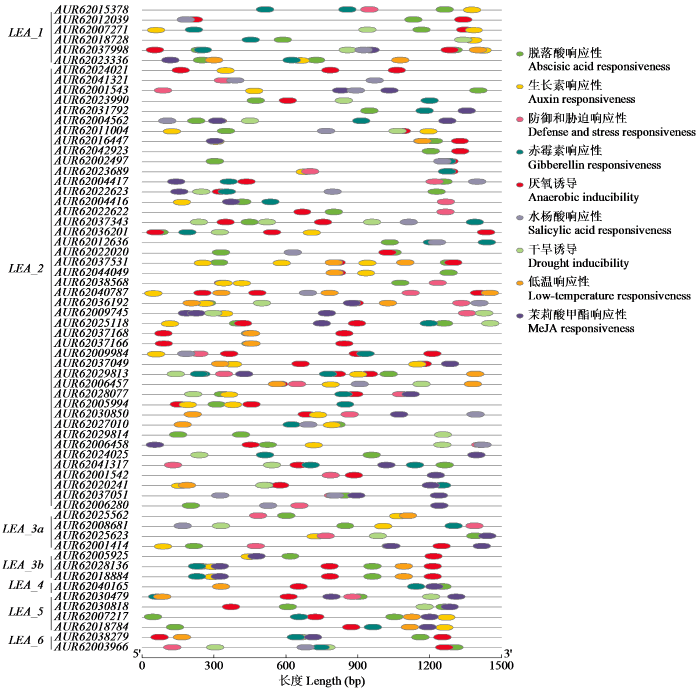
图6
藜麦LEA基因家族成员的顺式作用元件分析
Fig.6
Cis-acting element analysis in promoter of LEA gene family members
2.6 藜麦LEA基因家族成员在盐胁迫条件下的表达丰度及GO注释
图7
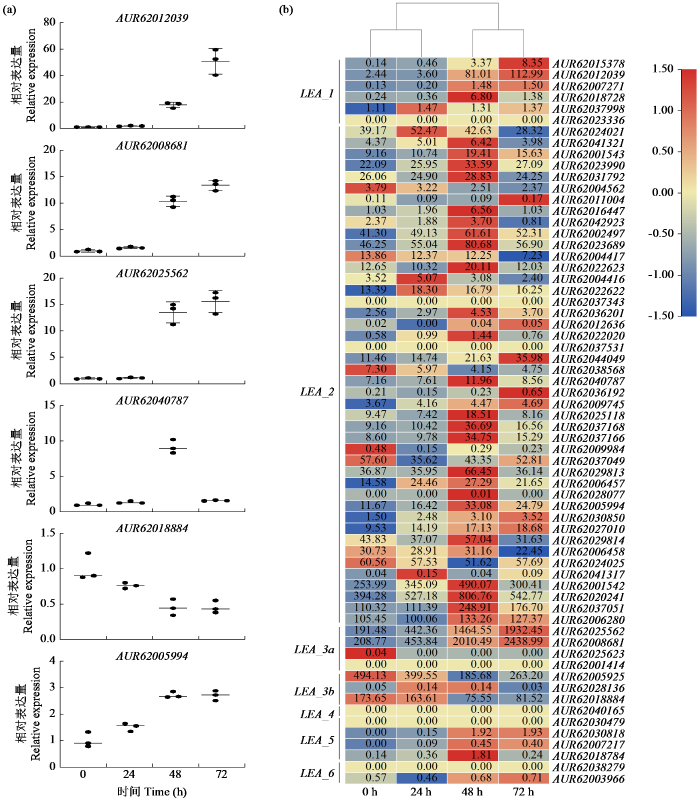
图7
盐胁迫条件下藜麦LEA基因家族成员的表达模式
Fig.7
Expression patterns of LEA gene family members in quinoa under salt stress
图8
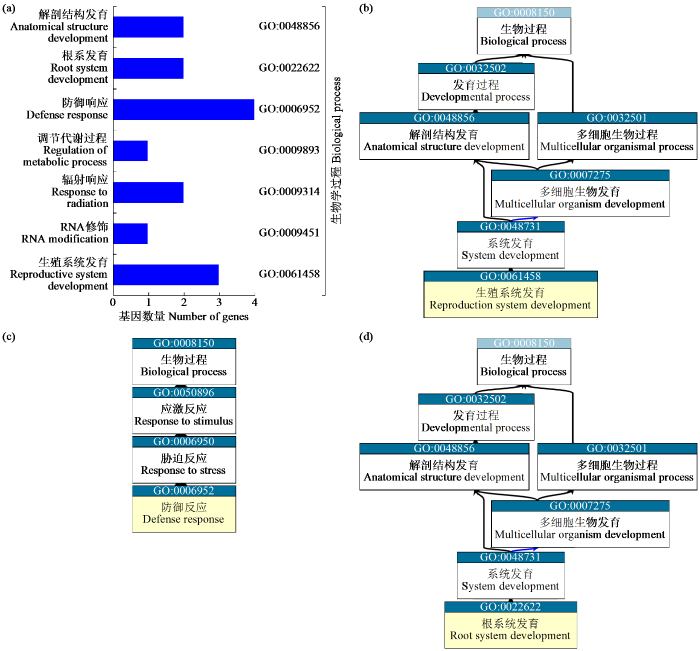
值得注意的是,LEA_1亚组中5个LEA基因表达随盐胁迫时间增加而提升,通过GO注释发现AUR62007271和AUR62018728参与生殖系统发育(GO:0061458)(图8b)。LEA_2亚组中存在31个LEA基因的表达随盐胁迫时间增加而提升,6个随盐胁迫时间增加而受到抑制,通过GO注释发现有4个LEA基因(AUR62025118、AUR62037168、AUR62037166和AUR62005994)参与防御反应(GO:0006952)(图8c)。LEA_3a亚组中AUR62025562和AUR62008681随盐胁迫时间增加而提升,并且参与根系发育(GO:0022622)(图8d)。LEA_3b亚组中的AUR62005925和AUR62018884随盐胁迫时间增加而受到抑制,LEA_4、LEA_5和LEA_6亚组并未发现有LEA基因随盐胁迫时间增加发生显著变化。
3 讨论
藜麦因较强的生境适应性受到广泛关注,鉴定其逆境应答基因可以为藜麦的抗逆性遗传改良提供靶向基因数据[17]。LEA蛋白是一类亲水性氨基酸占比较高的惰性蛋白,在作物响应非生物胁迫和增强逆境抗性中发挥着重要的作用[18]。本研究中,共鉴定出64个LEA基因,编码64个LEA蛋白,分属7个亚组。其中LEA_2亚组数量最多,80%以上的LEA蛋白为亲水性,与多数物种一致[5,9,15,19]。此外,LEA_2和LEA_3a亚组多分布于叶绿体,其它亚组均分布于细胞核,预示着不同亚组发挥的功能可能存在差异。值得注意的是,除LEA_2亚组中的AUR62022020含有8个内含子外,其余LEA基因内含子数量较少,这也表明较少的内含子可以快速调控LEA基因的表达,提升对环境的应激速率,同时也能减少转录过程中的能量损耗[20]。而motif基序显示,同一亚组之间具有相似的结构,不同亚组之间存在显著差异,如LEA_3a和LEA_3b亚组仅含有motif 9,LEA_5亚组仅含有motif 6,这可能与LEA蛋白特异性识别以及特定配体结合相关[21]。根据基因结构和蛋白基序的组内相似性和组间差异性,可以推测藜麦不同LEAs亚组间可能有不同的起源和进化方向[5,22]。除此之外,还发现LEA基因启动子区域除了含有各种激素调控元件以外,还含有TC-rich、LTR和MBS[23]等与非生物胁迫相关的顺势作用元件。推测LEA基因可能参与多种生物学过程,响应非生物胁迫。在盐胁迫条件下,藜麦萌发期的转录组数据中发现70%左右的LEA表达受到盐胁迫的诱导,主要分属于LEA_1、LEA_2、LEA_3a和LEA_3b亚组。但是通过GO注释发现,不同亚组参与不同的生物学过程,这表明LEA基因在藜麦盐胁迫过程中具有复杂的调控机制,且不同亚组的响应机制存在差异,其系统进化分析也支持了这一点。其中,LEA_2亚组主要参与植物的防御反应(GO:0006952),在低温、干旱、盐碱和ABA处理等非生物胁迫后,LEA_2蛋白迅速响应应激反应并大量表达,其带负电区域可以结合金属离子,减少氧化应激[24],亲水结构(K、Y和S片段)可以形成两亲性α螺旋,提升膜蛋白互作能力,维持膜完整性和流动性,减轻氧化损伤[25-26]。
在鉴定出的64个LEA基因中,发现11对同源基因的Ka/Ks均小于1,且同源基因对均在同一亚组内,这意味着各亚组进化具有较高的保守型,组内功能分化有限,以维持原始功能为主。9对串联重复基因Ka/Ks同样均小于1,仅存在于LEA_2亚组,预示着以片段重复为主的基因复制事件是LEA_2亚组扩增的主要动力,其结构稳定性和功能保守性可以减轻有害突变积累,从而保证LEA_2亚组在净化选择压力下维持核心保护功能[27]。物种间共线性分析发现藜麦与双子叶植物大豆、蒺藜苜蓿、拟南芥具有较多的共线性基因对,且呈现成簇共线性分布,表明这些LEA基因对可能源于双子叶共同祖先的串联重复或基因组加倍事件;与单子叶植物水稻共线性较少,主要以碎片化形式存在,反映了LEA仅基础核心功能基因在双子叶植物与单子叶植物物种间保持保守性。
4 结论
本研究在藜麦全基因水平上,鉴定了64个LEA基因,编码64个LEA蛋白,同一亚组具有相似的基因结构和保守基序。基因复制事件是LEA基因家族成员扩增的主要动力,在净化选择压力下维持了其核心功能,且与豆科植物进化具有较高的保守型。启动子区域存在多种激素和非生物胁迫相关的作用元件,通过转录组数据发现46个LEA基因受到盐胁迫诱导表达,GO注释显示主要参与结构发育、根系发育、防御响应和生殖系统发育等生物学过程。
参考文献
Overexpression of rice OsLEA5 relieves the deterioration in seed quality caused by high- temperature stress
DOI:10.5511/plantbiotechnology.21.0603a
PMID:34782824
[本文引用: 1]

Late embryogenesis abundant protein (LEA) genes are widely conserved in seed plant species and form a multigene family. While some LEAs are known to respond to environmental stresses, the function of many LEAs is unknown. OsLEA5 (Lea14A) interacts with a regulator of the endosperm storage production, FLO2, suggesting that OsLEA5 may be involved in endosperm quality control. RNAi knockdown line of showed decreased seed weight. Transformant lines overexpressing exhibited improved quality and seed weight of mature seeds when they were developed under high-temperature conditions, while seed quality strongly declined in wild-type plants exposed to high-temperature stress. These findings indicate that contributes to suppressing the deterioration of seed quality when developed under high-temperature conditions.© 2021 Japanese Society for Plant Biotechnology.
Desiccation tolerance in Ramonda serbica panc.: an integrative transcriptomic, proteomic, metabolite and photosynthetic study
DOI:10.3390/plants11091199
URL
[本文引用: 1]
The resurrection plant Ramonda serbica Panc. survives long desiccation periods and fully recovers metabolic functions within one day upon watering. This study aimed to identify key candidates and pathways involved in desiccation tolerance in R. serbica. We combined differential transcriptomics and proteomics, phenolic and sugar analysis, FTIR analysis of the cell wall polymers, and detailed analysis of the photosynthetic electron transport (PET) chain. The proteomic analysis allowed the relative quantification of 1192 different protein groups, of which 408 were differentially abundant between hydrated (HL) and desiccated leaves (DL). Almost all differentially abundant proteins related to photosynthetic processes were less abundant, while chlorophyll fluorescence measurements implied shifting from linear PET to cyclic electron transport (CET). The levels of H2O2 scavenging enzymes, ascorbate-glutathione cycle components, catalases, peroxiredoxins, Fe-, and Mn superoxide dismutase (SOD) were reduced in DL. However, six germin-like proteins (GLPs), four Cu/ZnSOD isoforms, three polyphenol oxidases, and 22 late embryogenesis abundant proteins (LEAPs; mainly LEA4 and dehydrins), were desiccation-inducible. Desiccation provoked cell wall remodeling related to GLP-derived H2O2/HO● activity and pectin demethylesterification. This comprehensive study contributes to understanding the role and regulation of the main metabolic pathways during desiccation aiming at crop drought tolerance improvement.
Developmental biochemistry of cottonseed embryogenesis and germination: changing messenger ribonucleic acid populations as shown by in vitro and in vivo protein synthesis
DOI:10.1021/bi00517a033
PMID:7284317
[本文引用: 1]
Changes in messenger ribonucleic acid (mRNA) populations during embryogenesis of cottonseed have been followed by cataloging (a) extant proteins, (b) proteins synthesized in vivo, and (c) proteins synthesized in vitro from extracted RNA, all at specific stages of embryogenesis. Evidence is presented for the existence of five mRNA subsets, all apparently under different regulatory regimes, that produce the abundant proteins of embryogenesis. One of these functions principally during the cell division phase of embryogenesis and encodes among its products the seed storage proteins whose mRNA is superabundant during this period. This subset has disappeared from the abundant group by the mature seed stage. Two other subsets appear in late embryogenesis, one of which may result from the removal of the embryo from the maternal environment, since it is inducible by excision of the young embryo from the seed. The other appears to be induced by the plant growth regulator abscisic acid, whose endogenous concentration increases at this stage. It can be induced by incubating excised young embryos in abscisic acid. The last two subsets exist throughout embryogenesis, but only one of them appears to function in germination.
Genome-wide identification,evolutionary and expression analyses of LEA gene family in peanut (Arachis hypogaea L.)
DOI:10.1186/s12870-022-03462-7
[本文引用: 1]
\n Late embryogenesis abundant (LEA) proteins are a group of highly hydrophilic glycine-rich proteins, which accumulate in the late stage of seed maturation and are associated with many abiotic stresses. However, few peanut\n LEA\n genes had been reported, and the research on the number, location, structure, molecular phylogeny and expression of\n AhLEA\n s was very limited.\n
The LEA gene family in tomato and its wild relatives: genome-wide identification, structural characterization, expression profiling, and role of SlLEA6 in drought stress
DOI:10.1186/s12870-022-03953-7
[本文引用: 4]
Late embryogenesis abundant (LEA) proteins are widely distributed in higher plants and play crucial roles in regulating plant growth and development processes and resisting abiotic stress. Cultivated tomato (Solanum lycopersicum) is an important vegetable crop worldwide; however, its growth, development, yield, and quality are currently severely constrained by abiotic stressors. In contrast, wild tomato species are more tolerant to abiotic stress and can grow normally in extreme environments. The main objective of this study was to identify, characterize, and perform gene expression analysis of LEA protein families from cultivated and wild tomato species to mine candidate genes and determine their potential role in abiotic stress tolerance in tomatoes.
The in vitro structure and functions of the disordered late embryogenesis abundant three proteins
DOI:10.1002/pro.4028
PMID:33474748
[本文引用: 1]
Late embryogenesis abundant (LEA) proteins are produced during seed embryogenesis and in vegetative tissue in response to various abiotic stressors. A correlation has been established between LEA expression and stress tolerance, yet their precise biochemical mechanism remains elusive. LEA proteins are very rich in hydrophilic amino acids, and they have been found to be intrinsically disordered proteins (IDPs) in vitro. Here, we perform biochemical and structural analyses of the four LEA3 proteins from Arabidopsis thaliana (AtLEA3). We show that the LEA3 proteins are disordered in solution but have regions with propensity for order. All LEA3 proteins were effective cryoprotectants of LDH in the freeze/thaw assays, while only one member, AtLEA3-4, was shown to bind Cu and Fe ions with micromolar affinity. As well, only AtLEA3-4 showed binding and a gain in α-helicity in the presence of the membrane mimic dodecylphosphocholine (DPC). We explored this interaction in greater detail using N-heteronuclear single quantum coherence (HSQC) nuclear magnetic resonance, and demonstrate that two sets of conserved motifs present in AtLEA3-4 are involved in the interaction with the DPC micelles, which themselves gain α-helical structure.© 2021 The Protein Society.
Group 3 LEA protein model peptides protect enzymes against desiccation stress
ROS homeostasis in abiotic stress tolerance in plants
DOI:10.3390/ijms21155208
URL
[本文引用: 1]
Climate change-induced abiotic stress results in crop yield and production losses. These stresses result in changes at the physiological and molecular level that affect the development and growth of the plant. Reactive oxygen species (ROS) is formed at high levels due to abiotic stress within different organelles, leading to cellular damage. Plants have evolved mechanisms to control the production and scavenging of ROS through enzymatic and non-enzymatic antioxidative processes. However, ROS has a dual function in abiotic stresses where, at high levels, they are toxic to cells while the same molecule can function as a signal transducer that activates a local and systemic plant defense response against stress. The effects, perception, signaling, and activation of ROS and their antioxidative responses are elaborated in this review. This review aims to provide a purview of processes involved in ROS homeostasis in plants and to identify genes that are triggered in response to abiotic-induced oxidative stress. This review articulates the importance of these genes and pathways in understanding the mechanism of resistance in plants and the importance of this information in breeding and genetically developing crops for resistance against abiotic stress in plants.
Genome-wide identification of late embryogenesis abundant protein family and their key regulatory network in Pinus tabuliformis cold acclimation
DOI:10.1093/treephys/tpad095 URL [本文引用: 2]
Characterization of a novel TtLEA2 gene from Tritipyrum and its transformation in wheat to enhance salt tolerance
DOI:10.3389/fpls.2022.830848 URL [本文引用: 1]
Genome-wide search and structural and functional analyses for late embryogenesis- abundant (LEA) gene family in poplar
DOI:10.1186/s12870-021-02872-3
[本文引用: 1]
The Late Embryogenesis-Abundant (LEA) gene families, which play significant roles in regulation of tolerance to abiotic stresses, widely exist in higher plants. Poplar is a tree species that has important ecological and economic values. But systematic studies on the gene family have not been reported yet in poplar.
Quinoa abiotic stress responses: a review
DOI:10.3390/plants7040106 URL [本文引用: 1]
Exploring geographic variations in quinoa grains: unveiling anti-alzheimer activity via GC-MS, LC-QTOF-MS/MS, molecular networking, and chemometric analysis
DOI:10.1016/j.foodchem.2024.141918 URL [本文引用: 1]
The genome of Chenopodium quinoa
DOI:10.1038/nature21370
[本文引用: 1]
\n Chenopodium quinoa (quinoa) is a highly nutritious grain identified as an important crop to improve world food security. Unfortunately, few resources are available to facilitate its genetic improvement. Here we report the assembly of a high-quality, chromosome-scale reference genome sequence for quinoa, which was produced using single-molecule real-time sequencing in combination with optical, chromosome-contact and genetic maps. We also report the sequencing of two diploids from the ancestral gene pools of quinoa, which enables the identification of sub-genomes in quinoa, and reduced-coverage genome sequences for 22 other samples of the allotetraploid goosefoot complex. The genome sequence facilitated the identification of the transcription factor likely to control the production of anti-nutritional triterpenoid saponins found in quinoa seeds, including a mutation that appears to cause alternative splicing and a premature stop codon in sweet quinoa strains. These genomic resources are an important first step towards the genetic improvement of quinoa.
Identification and expression analysis of LEA gene family members in pepper (Capsicum annuum L.)
DOI:10.1002/feb4.v13.12 URL [本文引用: 2]
Genome-wide identification of polyamine metabolism and ethylene synthesis genes in Chenopodium quinoa Willd. and their responses to low-temperature stress
DOI:10.1186/s12864-024-10265-7
[本文引用: 1]
Quinoa (Chenopodium quinoa Willd.) is valued for its nutritional richness. However, pre-harvest sprouting poses a significant threat to yield and grain quality. This study aims to enhance our understanding of pre-harvest sprouting mitigation strategies, specifically through delayed sowing and avoiding rainy seasons during quinoa maturation. The overarching goal is to identify cold-resistant varieties and unravel the molecular mechanisms behind the low-temperature response of quinoa. We employed bioinformatics and genomics tools for a comprehensive genome-wide analysis of polyamines (PAs) and ethylene synthesis gene families in quinoa under low-temperature stress.
Genome-wide identification of the LEA gene family in Panax ginseng: evidence for the role of PgLEA2-50 in plant abiotic stress response
DOI:10.1016/j.plaphy.2024.108742 URL [本文引用: 1]
The role of the late embryogenesis-abundant (LEA) protein family in development and the abiotic stress response: a comprehensive expression analysis of potato (Solanum tuberosum)
DOI:10.3390/genes10020148
URL
[本文引用: 1]
Late embryogenesis-abundant (LEA) proteins are a large and highly diverse family believed to function in normal plant growth and development, and in protecting cells from abiotic stress. This study presents a characterisation of 74 Solanum tuberosum LEA (StLEA) proteins belonging to nine groups. StLEA genes have few introns (≤2) and are distributed on all chromosomes, occurring as gene clusters on chromosomes 1, 2, and 10. All four StASR (StLEA7 group) genes were concentrated on chromosome 4, suggesting their evolutionary conservation on one chromosome. Expression profiles of StLEA genes, in different tissues and in response to hormone and stress treatments, indicated that 71 StLEA genes had differential expression levels, of which 68 StLEA genes were differentially expressed in response to hormones and stress exposure in the potato. Continuous high expression of StASR-2, StLEA3-3, StDHN-3, StLEA2-29, and StLEA2-14 in different tissues indicated their contribution to plant development processes. StLEA2-14, StLEA2-31, StLEA3-3, StASR-1, and StDHN-1 were upregulated by six abiotic stresses, showing their tolerance to a wide spectrum of environmental stresses. Expression analysis of 17 selected StLEA genes in response to drought, salt, heavy metal, heat, and cold treatments by quantitative real-time polymerase chain reaction indicated that StLEA proteins may be involved in distinct signalling pathways. Taken together, StLEA3, StDHN, and StASR subgroup genes may be excellent resources for potato defence against environmental stresses. These results provide valuable information and robust candidate genes for future functional analysis aimed at improving the stress tolerance of the potato.
Comparative analysis of the exon- intron structure in eukaryotic genomes
DOI:10.4236/ym.2017.11006 URL [本文引用: 1]
LEA motifs promote desiccation tolerance in vivo
DOI:10.1186/s12915-021-01176-0
PMID:34903234
[本文引用: 1]
Cells and organisms typically cannot survive in the absence of water. However, some animals including nematodes, tardigrades, rotifers, and some arthropods are able to survive near-complete desiccation. One class of proteins known to play a role in desiccation tolerance is the late embryogenesis abundant (LEA) proteins. These largely disordered proteins protect plants and animals from desiccation. A multitude of studies have characterized stress-protective capabilities of LEA proteins in vitro and in heterologous systems. However, the extent to which LEA proteins exhibit such functions in vivo, in their native contexts in animals, is unclear. Furthermore, little is known about the distribution of LEA proteins in multicellular organisms or tissue-specific requirements in conferring stress protection. Here, we used the nematode C. elegans as a model to study the endogenous function of an LEA protein in an animal.We created a null mutant of C. elegans LEA-1, as well as endogenous fluorescent reporters of the protein. LEA-1 mutant animals formed defective dauer larvae at high temperature. We confirmed that C. elegans lacking LEA-1 are sensitive to desiccation. LEA-1 mutants were also sensitive to heat and osmotic stress and were prone to protein aggregation. During desiccation, LEA-1 expression increased and became more widespread throughout the body. LEA-1 was required at high levels in body wall muscle for animals to survive desiccation and osmotic stress, but expression in body wall muscle alone was not sufficient for stress resistance, indicating a likely requirement in multiple tissues. We identified minimal motifs within C. elegans LEA-1 that were sufficient to increase desiccation survival of E. coli. To test whether such motifs are central to LEA-1's in vivo functions, we then replaced the sequence of lea-1 with these minimal motifs and found that C. elegans dauer larvae formed normally and survived osmotic stress and mild desiccation at the same levels as worms with the full-length protein.Our results provide insights into the endogenous functions and expression dynamics of an LEA protein in a multicellular animal. The results show that LEA-1 buffers animals from a broad range of stresses. Our identification of LEA motifs that can function in both bacteria and in a multicellular organism in vivo suggests the possibility of engineering LEA-1-derived peptides for optimized desiccation protection.© 2021. The Author(s).
Nuclear translocation of OsMFT 1 that is impeded by OsFTIP1 promotes drought tolerance in rice
DOI:10.1016/j.molp.2021.05.001 URL [本文引用: 1]
Genome-wide identification and expression analysis of glycine-rich RNA-binding protein family in sweet potato wild relative Ipomoea trifida
DOI:S0378-1119(18)31187-9
PMID:30453066
[本文引用: 1]
Glycine-rich RNA-binding proteins (GRPs) contain RNA recognition motif (RRM) and glycine-rich domains at the N- or C-terminus, respectively, and they participate in varied physiological and biochemical processes, as well as environmental stresses. Sweet potato from the genus Ipomoea is one of the most important crops. However, the role of the GRP gene family in Ipomoea plant species has not been reported yet. At the same time, the genome of sweet potato remains to be elucidated, but the genome of I. trifida which is most probably the progenitor of the sweet potato was released recently. In this regard, we carried out genome-wide analysis of GRP family members in I. trifida. Here, we identified nine GRP genes in I. trifida and investigated their motif distribution, promoters and gene structure. Subsequently, we performed phylogenetic analysis with the GRP genes from I. trifida, Arabidopsis thaliana, Zea mays L. and Oryza sativa to investigate their phylogenetic relationship. Moreover, we studied the expression patterns of ItGRPs in the roots, stems, young and mature leaves and flowers and found that ItGRP genes were tissue-specific. Meanwhile, the expression profiles under four abiotic stress conditions, including heat, cold, salt and drought stress treatments, revealed that some genes were markedly up-regulated or down-regulated. Taken together, our findings will provide reference to studies on the function of GRP genes in the development and stress response of I. trifida.Copyright © 2018 The Authors. Published by Elsevier B.V. All rights reserved.
Comparative analysis of the LEA gene family in seven Ipomoea species, focuses on sweet potato (Ipomoea batatas L.)
DOI:10.1186/s12870-024-05981-x
[本文引用: 1]
\n Late Embryogenesis Abundant (LEA) proteins are extensively distributed among higher plants and are crucial for regulating growth, development, and abiotic stress resistance. However, comprehensive data regarding the\n LEA\n gene family in\n Ipomoea\n species remains limited. In this study, we conducted a genome-wide comparative analysis across seven\n Ipomoea\n species, including sweet potato (\n I. batatas\n ),\n I. trifida\n,\n I. triloba\n,\n I. nil\n,\n I. purpurea\n,\n I. cairica\n, and\n I. aquatica\n, identifying 73, 64, 77, 62, 70, 70, and 74\n LEA\n genes, respectively. The\n LEA\n genes were divided into eight subgroups: LEA_1, LEA_2, LEA_3, LEA_4, LEA_5, LEA_6, SMP, and Dehydrin according to the classification of the\n LEA\n family in Arabidopsis. Gene structure and protein motif analyses revealed that genes within the same phylogenetic group exhibited comparable exon/intron structures and motif patterns. The distribution of\n LEA\n genes across chromosomes varied among the different\n Ipomoea\n species. Duplication analysis indicated that segmental and tandem duplications significantly contributed to the expansion of the\n LEA\n gene family, with segmental duplications being the predominant mechanism. The analysis of the non-synonymous (Ka) to synonymous (Ks) ratio (Ka/Ks) indicated that the duplicated\n Ipomoea LEA\n genes predominantly underwent purifying selection. Extensive cis-regulatory elements associated with stress responses were identified in the promoters of\n LEA\n genes. Expression analysis revealed that the\n LEA\n gene exhibited widespread expression across diverse tissues and showed responsive modulation to various abiotic stressors. Furthermore, we selected 15\n LEA\n genes from sweet potatoes for RT-qPCR analysis, demonstrating that five genes responded to salt stress in roots, while three genes were responsive to drought stress in leaves. Additionally, expression changes of seven genes varied at different stages of sweet potato tuber development. These findings enhanced our understanding of the evolutionary dynamics of\n LEA\n genes within the\n Ipomoea\n genome and may inform future molecular breeding strategies for sweet potatoes.\n
Genome-wide identification and characterization of members of the LEA gene family in Panax notoginseng and their transcriptional responses to dehydration of recalcitrant seeds
DOI:10.1186/s12864-023-09229-0
[本文引用: 1]
\n Late embryogenesis abundant (LEA) proteins play an important role in dehydration process of seed maturation. The seeds of\n Panax notoginseng\n (Burkill) F. H. Chen are typically characterized with the recalcitrance and are highly sensitive to dehydration. However, it is not very well known about the role of LEA proteins in response to dehydration stress in\n P. notoginseng\n seeds. We will perform a genome-wide analysis of the LEA gene family and their transcriptional responses to dehydration stress in recalcitrant\n P. notoginseng\n seeds.\n
Genome-wide identification and analysis of LEA_2 gene family in alfalfa (Medicago sativa L.) under aluminum stress
DOI:10.3389/fpls.2022.976160
URL
[本文引用: 1]
Late embryonic development abundant proteins (LEAs) are a large family of proteins commonly existing in plants. LEA_2 is the largest subfamily in the LEA, it plays an important role in plant resistance to abiotic stress. In order to explore the characteristics of LEA_2 gene family members in alfalfa (Medicago sativa L.), 155 members of LEA_2 (MsLEA_2) family were identified from alfalfa genome. Bioinformatics analysis was conducted from the aspects of phylogenetic relationship, chromosome distribution, chromosome colinearity, physical and chemical properties, motif composition, exon-intron structure, cis-element and so on. Expression profiles of MsLEA_2 gene were obtained based on Real-time fluorescent quantitative PCR (qRT-PCR) analysis and previous RNA-seq data under aluminum (Al) stress. Bioinformatics results were shown that the MsLEA_2 genes are distributed on all 32 chromosomes. Among them, 85 genes were present in the gene clusters, accounting for 54.83%, and chromosome Chr7.3 carries the largest number of MsLEA_2 (19 LEA_2 genes on Chr7.3). Chr7.3 has a unique structure of MsLEA_2 distribution, which reveals a possible special role of Chr7.3 in ensuring the function of MsLEA_2. Transcriptional structure analysis revealed that the number of exons in each gene varies from 1 to 3, and introns varies from 0 to 2. Cis-element analysis identified that the promoter region of MsLEA_2 is rich in ABRE, MBS, LTR, and MeJARE, indicating MsLEA_2 has stress resistance potential under abiotic stress. RNA-seq data and qRT-PCR analyses showed that most of the MsLEA_2 members were up-regulated when alfalfa exposed to Al stress. This study revealed that phylogenetic relationship and possible function of LEA_ 2 gene in alfalfa, which were helpful for the functional analysis of LEA_ 2 proteins in the future and provided a new theoretical basis for improving Al tolerance of alfalfa.
Demographic history and natural selection shape patterns of deleterious mutation load and barriers to introgression across Populus genome

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}