


豌豆(Pisum sativum L. 2n=14)有7对同源染色体,豌豆属自花授粉作物[1]。豌豆富含蛋白质、淀粉及人体所需的多种微量元素,营养均衡,是非常利于食用的杂粮作物,遗传多样性丰富,在我国被广泛种植[2-3]。目前,国家作物种质库保存了7000余份豌豆种质资源,占全球豌豆种质资源的29%,其中我国特有种质资源达71%[4]。由于不同地区间品种交流,导致来源较为复杂,存在大量“同名异种”和“一品多名”种质资源,许多生产者不能区分,无法保障豌豆产量和农民的收入[5]。因此,为确保豌豆优质品种的真实性,建立一套便捷、快速且可靠的豌豆良种鉴定技术,对于品种认定和保护育种工作者的知识产权具有重要意义。
简单重复序列(simple sequence repeat,SSR)一般是由1~6个不等的碱基对串联重复而成的DNA序列,又称为微卫星DNA(microsatellite DNA)[6-
电子PCR(electronic PCR,e-PCR)是一种核苷酸序列电子分析工具,在DNA片段的染色体定位、基因组辅助作图、PCR引物辅助设计和基因克隆等方面发挥着重要作用[8,20]。熊登坤等[14]用PCR验证结果表明利用电子PCR可以提高SSR多态性引物的筛选效率。陆景标[21]通过e-PCR验证设计的引物,评估得出高丹草编码区SSR位点的多态性高于非编码区,证明通过e-PCR可以快速筛选引物,并可以验证其是否具有多态性。管培丽等[22]通过e-PCR对已发表的芸薹属作物SSR标记进行分析,并通过真实PCR验证,发现准确率高达80.56%,证明e-PCR技术显著提高标记开发的准确性和可预见性,节省了大量的标记验证成本,为标记的后期高效利用奠定了基础。
本研究利用SSRMMD(simple sequence repeat molecular marker developer,SSRMMD)软件对豌豆基因组数据扫描获得SSR位点,设计SSR引物。运用TBtools中的e-PCR功能对发布的豌豆品种v1a、中豌六号、豌豆07v1和豌豆87v1基因组进行电子PCR-SSR分子标记多态性分析,以提高SSR分子标记开发效率和多态性引物筛选效率,再利用SSR分子标记对收集到的32份豌豆种质资源进行分析,构建了其SSR指纹图谱,为豌豆种质资源的鉴定与评价、资源保护及育种应用提供了理论依据。
1 材料与方法
1.1 试验材料
豌豆种质资源来自于甘肃、江苏、山东、宁夏、河北、广东深圳和重庆的地方品种,共32份(表1),由黑龙江八一农垦大学生命科学技术学院植物发育生物学实验室保存。
表1 不同品种豌豆引种地点
Table 1
| 编号Code | 品种Variety | 引种地点Introduction site |
|---|---|---|
| 1 | DWD05-01 | 甘肃 |
| 2 | 成豌11号 | 江苏 |
| 3 | c13 | 重庆 |
| 4 | 方绿豌豆 | 山东 |
| 5 | 杂粮甜豌豆 | 江苏 |
| 6 | zx-wd-0030 | 甘肃 |
| 7 | 定豌7号 | 甘肃 |
| 8 | zx-wd-0007 | 甘肃 |
| 9 | zx-wd-0013 | 甘肃 |
| 10 | 麻豌豆 | 山东 |
| 11 | 澳洲红玉豌豆 | 山东 |
| 12 | S6071 | 甘肃 |
| 13 | 黑眼豌豆 | 山东 |
| 14 | SW1102 | 甘肃 |
| 15 | c01 | 重庆 |
| 16 | 1702 | 甘肃 |
| 17 | 彩豌豆 | 山东 |
| 18 | S5008 | 甘肃 |
| 19 | RC09 | 甘肃 |
| 20 | 食荚型甜豌豆 | 山东 |
| 21 | 鲜嫩二号豌豆 | 宁夏 |
| 22 | 美国甜脆豌豆 | 宁夏 |
| 23 | 豌豆 | 山东 |
| 24 | 德利荷兰豆 | 河北 |
| 25 | zx-wd-0019 | 甘肃 |
| 26 | 优选法国008香豆 | 深圳 |
| 27 | DWD05-03 | 甘肃 |
| 28 | 猪耳朵 | 山东 |
| 29 | zx-wd-0009 | 甘肃 |
| 30 | DWD05-10 | 甘肃 |
| 31 | WD04-09 | 甘肃 |
| 32 | 黑豌豆 | 山东 |
1.2 试验方法
1.2.1 田间表型性状调查及DNA提取
供试材料播种于黑龙江省绥化市安达市黑龙江八一农垦大学试验田;土壤为黑钙土,碱解氮96.10 mg/kg,有效磷6.32 mg/kg,速效钾72.90 mg/kg。播种时间为2020年5月10日;垄距65 cm,株距5 cm;播种深度3~5 cm。记录成熟期、株高、节数、荚长、单荚粒数、荚型、粒色、粒形和百粒重共9个性状。苗期取豌豆幼嫩叶片0.2~0.4 g,采用CTAB方法提取其总DNA[23],用适量TE缓冲液溶解后,储存在-20 ℃备用。
1.2.2 SSR引物标记开发与SSR引物设计
豌豆基因组序列来源于NCBI数据库(
1.2.3 e-PCR分析
利用TBtools中的e-PCR功能输入4个豌豆参考基因组“Pisum_ sativum_v1a”、“CAAS_Psat_ZW6_1.0”、“ASM3678607v1”和“ASM2453087v1”染色体序列的FASTA格式,将设计的SSR引物按FASTA格式输入,利用SSR引物在豌豆全基因组序列中进行匹配,设置输出文件,通过Excel对电子PCR的扩增产物长度、特异性和多态性进行分析。
1.2.4 PCR扩增及电泳检测
SSR引物由上海生工生物有限公司合成,PCR反应体系为20 μL:模板DNA 1 μL,正、反向引物10 pmo1/L各0.5 μL,2×Taq Master Mix 10 μL,dd H2O 8 μL。PCR扩增程序为95 ℃预变性5 min;95 ℃变性30 s,57 ℃退火30 s,72 ℃延伸45 s,共35个循环;72 ℃延伸5 min,16 ℃保存[24]。扩增产物采用6%非变性聚丙烯酰胺凝胶电泳检测,电泳在LF-CZ05A高通量垂直电泳槽中进行。利用Gel-Red固定20 min显色,放入凝胶成像(Bio-Red)系统中照相记录。
1.2.5 数据分析
人工读取电泳凝胶上SSR分离条带所显示的相对位置信息并通过Image Lab软件对不明显的条带进行校正,对SSR标记扩增产物进行统计。根据标准DNA分子量和引物的迁移率记录电泳结果,每对引物迁移率最大的条带记为1,迁移率次之的记为2,依次类推,无条带记为0,建立SSR基因型信息数据,按照引物顺序进行编码,得到的指纹代码即为该品种的特征指纹图谱[5]。
2 结果与分析
2.1 豌豆农艺性状分析
表2 不同品种豌豆农艺性状
Table 2
| 编号 Code | 品种 Variety | 花期 Flowering period | 花荚期 Flowering and pod-setting stage | 成熟期 Maturity | 荚型 Pod type | 粒色 Grain color | 粒形 Grain shape | 株高 Plant height (cm) | 节数 Number of sections | 荚长 Pod length (cm) | 单荚粒数 Number of grains per pod | 百粒重 100-grain weight (g) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | DWD05-01 | 06-24 | 07-08 | 08-27 | 硬 | 黄 | 皱 | 86 | 13 | 6.30 | 4.00 | 22.29 |
| 2 | 成豌11号 | 06-24 | 07-08 | 08-10 | 硬 | 黄 | 圆 | 50 | 14 | 6.60 | 5.67 | 22.03 |
| 3 | c13 | 07-01 | 07-15 | 08-01 | 硬 | 黄 | 皱 | 95 | 18 | 6.57 | 6.00 | 21.01 |
| 4 | 方绿豌豆 | 06-17 | 07-01 | 08-01 | 硬 | 绿 | 皱 | 25 | 12 | 6.67 | 5.33 | 38.86 |
| 5 | 杂粮甜豌豆 | 06-10 | 06-24 | 08-01 | 硬 | 绿 | 皱 | 45 | 13 | 6.73 | 5.67 | 25.17 |
| 6 | zx-wd-0030 | 07-01 | 07-15 | 08-15 | 硬 | 绿 | 皱 | 120 | 21 | 7.32 | 5.50 | 25.76 |
| 7 | 定豌7号 | 06-24 | 07-08 | 08-01 | 硬 | 黄 | 皱 | 130 | 16 | 6.53 | 6.33 | 25.31 |
| 8 | zx-wd-0007 | 06-03 | 06-17 | 07-25 | 硬 | 绿 | 皱 | 120 | 14 | 6.54 | 4.50 | 24.70 |
| 9 | zx-wd-0013 | 07-08 | 07-22 | 08-02 | 硬 | 黄 | 皱 | 110 | 13 | 5.50 | 4.33 | 21.52 |
| 10 | 麻豌豆 | 07-08 | 07-22 | 09-01 | 硬 | 麻 | 皱 | 50 | 18 | 5.87 | 4.33 | 19.92 |
| 11 | 澳洲红玉豌豆 | 06-24 | 07-08 | 08-05 | 硬 | 黄 | 皱 | 60 | 18 | 6.37 | 4.33 | 23.52 |
| 12 | S6071 | 06-24 | 07-08 | 08-01 | 硬 | 黄 | 皱 | 55 | 13 | 7.23 | 5.00 | 27.38 |
| 13 | 黑眼豌豆 | 06-17 | 07-01 | 08-01 | 硬 | 黄 | 皱 | 185 | 27 | 5.60 | 4.50 | 17.71 |
| 14 | SW1102 | 06-17 | 07-01 | 08-01 | 硬 | 黄 | 皱 | 50 | 16 | 5.60 | 4.00 | 27.40 |
| 15 | c01 | 06-17 | 07-01 | 08-01 | 硬 | 黄 | 皱 | 51 | 13 | 6.60 | 6.00 | 18.84 |
| 16 | 1702 | 06-17 | 07-01 | 08-05 | 硬 | 黄 | 皱 | 51 | 15 | 5.87 | 4.00 | 24.15 |
| 17 | 彩豌豆 | 06-10 | 06-24 | 08-01 | 硬 | 黄 | 皱 | 60 | 18 | 5.27 | 4.67 | 21.69 |
| 18 | S5008 | 06-24 | 07-08 | 08-05 | 硬 | 绿 | 皱 | 60 | 20 | 7.47 | 5.33 | 30.69 |
| 19 | RC09 | 06-17 | 07-01 | 08-01 | 硬 | 麻 | 圆 | 85 | 16 | 6.24 | 5.62 | 26.07 |
| 20 | 食荚型甜豌豆 | 06-17 | 07-01 | 08-01 | 硬 | 绿 | 皱 | 55 | 14 | 6.58 | 4.78 | 18.71 |
| 21 | 鲜嫩二号豌豆 | 06-24 | 07-08 | 08-17 | 硬 | 黄 | 皱 | 80 | 17 | 7.27 | 7.33 | 22.14 |
| 22 | 美国甜脆豌豆 | 06-24 | 07-08 | 07-15 | 硬 | 绿 | 皱 | 135 | 28 | 6.97 | 6.33 | 17.86 |
| 23 | 豌豆 | 06-17 | 07-01 | 08-05 | 硬 | 绿 | 皱 | 110 | 32 | 6.57 | 3.00 | 21.05 |
| 24 | 德利荷兰豆 | 06-24 | 07-08 | 08-01 | 硬 | 绿 | 皱 | 55 | 16 | 8.07 | 8.00 | 22.03 |
| 25 | zx-wd-0019 | 07-01 | 07-15 | 08-15 | 硬 | 黄 | 皱 | 61 | 12 | 5.90 | 4.00 | 24.27 |
| 26 | 优选法国008香豆 | 06-24 | 07-08 | 08-01 | 硬 | 绿 | 皱 | 70 | 11 | 6.67 | 4.67 | 26.97 |
| 27 | DWD05-03 | 06-17 | 07-01 | 08-17 | 硬 | 绿 | 皱 | 170 | 34 | 5.67 | 3.00 | 18.31 |
| 28 | 猪耳朵 | 07-08 | 07-22 | 08-25 | 硬 | 黄 | 皱 | 125 | 18 | 5.23 | 3.00 | 29.18 |
| 29 | zx-wd-0009 | 07-01 | 07-15 | 08-15 | 硬 | 黄 | 皱 | 60 | 18 | 5.63 | 3.67 | 23.07 |
| 30 | DWD05-10 | 07-08 | 07-22 | 08-15 | 硬 | 绿 | 皱 | 95 | 17 | 7.97 | 4.33 | 29.01 |
| 31 | WD04-09 | 06-17 | 07-01 | 08-10 | 硬 | 黄 | 皱 | 41 | 13 | 6.07 | 5.00 | 30.22 |
| 32 | 黑豌豆 | 07-01 | 07-15 | 08-10 | 硬 | 麻 | 皱 | 95 | 15 | 5.67 | 6.67 | 15.91 |
2.2 豌豆全基因组SSR频率及总体分布特征
通过SSRMMD软件对豌豆v1a全基因组序列中的SSR位点进行鉴定,获得311 601个SSR位点。其中二核苷酸重复单元占比最高,占67.25%;三、四核苷酸重复单元分别占总SSR位点的23.26%和9.49%(图1a)。二核苷酸重复单元中AT/TA(30.76%)和TA/AT(29.42%)占比较高,其中GC/CG和 CG/GC占比不足1.00%,可能为稀有SSR位点的主要成员。三核苷酸、四核苷酸虽然占比较少,但碱基组合的随机性和可选择性更强,种类更加多样,且重复单元占自身核苷酸重复类型的比例分布更加均匀。在双碱基和多碱基重复中,A、T碱基组合占比最高,因此豌豆全基因组SSR位点具有A/T丰富的特性,且随着A、T碱基组合数增加其所占比例呈下降趋势(图1b)。豌豆SSR序列长度在12~9960 bp,主要以12~44 bp短重复单元为主(92.45%),表明不同长度的重复单元占比存在较大差异,其中20 bp(15.38%)的SSR序列占比最高,并随着SSR序列长度增加,所占比例逐渐下降(图1c)。
图1
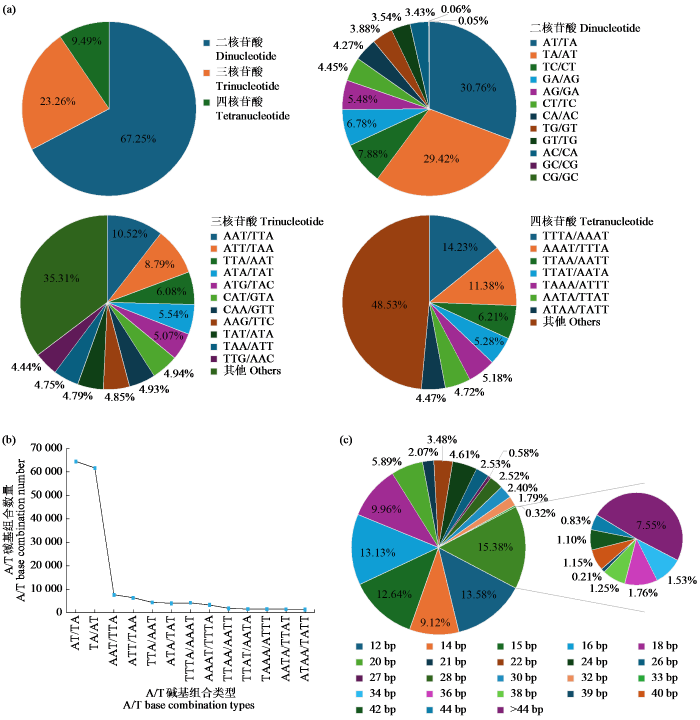
图1
豌豆全基因组SSR位点重复型的分布特征
(a) SSR位点中不同类型重复单元的占比,(b) 不同A/T碱基组合类型SSR位点数量分布,(c) SSR序列长度分布。
Fig.1
Distribution characteristics of SSR locus repeat types in whole pea genome
(a) the proportion of different types of repeat units in SSR locus, (b) the number distribution of SSR locus for different A/T base combination types, (c) SSR sequence length distribution.
2.3 豌豆基因组SSR标记及e-PCR分析
由表3可知,通过SSRMMD软件在豌豆v1a全基因组序列311 601个SSR位点中共获取165 288对SSR标记,豌豆v1a中SSR标记发生的平均频率为50.92对/Mb,平均21.20 kb有1个SSR标记;每条染色体上平均50.92对/Mb,其中Ps05染色体SSR标记最多(32 571),Ps04染色体SSR标记最少(11 618);Ps01染色体SSR标记密度最高(63.90对/Mb),Ps04染色体最低(26.02对/Mb)。
表3 豌豆基因组“Pisum sativum v1a”SSR标记分布
Table 3
| 染色体 Chromosome | 染色体大小 Chromosome size (Mb) | 标记数量 Number of markers | 标记密度(对/Mb) Marker density (pair/Mb) | 相邻两个SSR位点的平均距离 Average distance between two adjacent SSR locus (kb) |
|---|---|---|---|---|
| Ps01 | 372.17 | 23 784 | 63.90 | 15.65 |
| Ps02 | 427.60 | 22 370 | 52.32 | 19.11 |
| Ps03 | 437.56 | 20 142 | 46.03 | 21.72 |
| Ps04 | 446.35 | 11 618 | 26.02 | 38.43 |
| Ps05 | 579.30 | 32 571 | 56.22 | 17.79 |
| Ps06 | 480.42 | 27 080 | 56.36 | 17.74 |
| Ps07 | 497.38 | 27 663 | 55.62 | 17.98 |
| 平均Average | 462.97 | 23 604 | 50.92 | 21.20 |
在豌豆每条染色体中的SSR标记中,按比例挑选12~21对SSR标记进行e-PCR分析,共挑选122对SSR引物。结果发现122对SSR引物在豌豆v1a基因组中有110对能生成扩增产物,概率高达90.16%;在中豌六号基因组中有91对能生成扩增产物,概率为74.59%;在豌豆07v1基因组中有88对能生成扩增产物,概率为72.13%;在豌豆87v1基因组中有91对能生成扩增产物,概率为74.59%。
图2
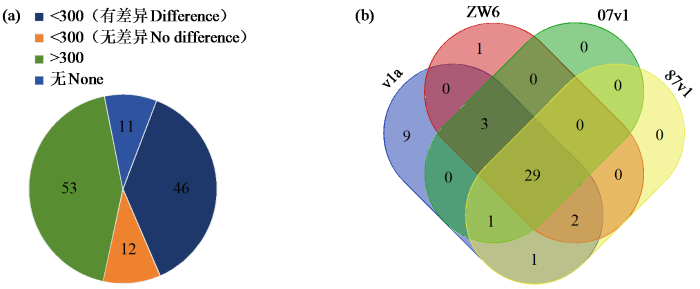
图2
4个豌豆基因组中有扩增产物的引物和扩增产物在300 bp以下的扩增数分析
(a) 4个豌豆基因组中不同扩增片段大小的个数,(b) 4个豌豆基因组中扩增产物在300 bp以下的维恩图,ZW6:中豌六号。
Fig.2
Analysis of primers with amplification products in four pea genomes and amplification number of amplification products below 300 bp
(a) The number of different amplified fragment sizes in four pea genomes, (b) Venn plots of amplification products below 300 bp in four pea genomes, ZW6: Zhongwan No.6.
图3
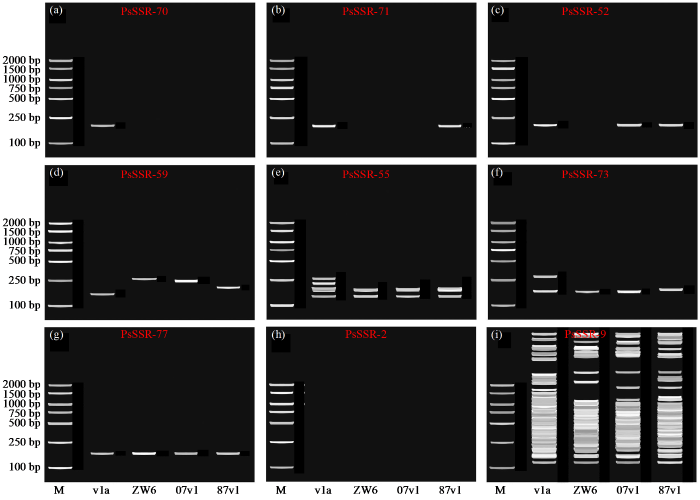
图3
部分SSR引物在4个豌豆基因组的e-PCR扩增情况
Fig.3
e-PCR amplification of some SSR primers in four pea genomes
2.4 SSR标记多态性分析
图4
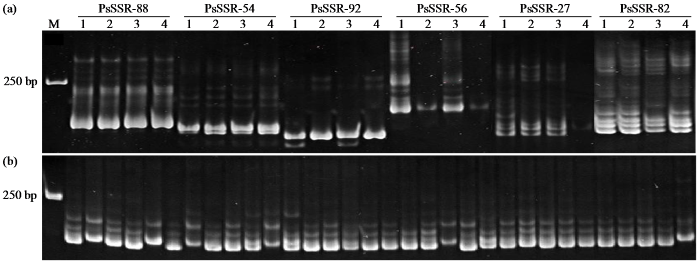
图4
部分SSR引物的PCR结果
(a) 部分SSR引物的PCR扩增产物,1:DWD05-01、2:成豌11号、3:方绿豌豆、4:杂粮甜豌豆。(b) PsSSR-82对32个豌豆品种的电泳检测结果。
Fig.4
PCR results of some SSR primers
(a) PCR amplification products of some SSR primers, 1: DWD05-01, 2: Cheng Pea No.11, 3: Square green pea, 4: mixed grain sweet peas. (b) electrophoretic detection results of PsSSR-82 on 32 pea varieties.
表4 SSR多态性检测结果
Table 4
| 编号Code | 引物类型Primer type | 引物总数Total number of primers | 多态性Polymorphism | 比例Proportion (%) |
|---|---|---|---|---|
| 1 | 扩增片段均小于300 bp(有差异,共显性标记) | 10 | 6 | 60 |
| 2 | 扩增片段均小于300 bp(有差异,显性标记) | 10 | 1 | 10 |
| 3 | 扩增片段均小于300 bp(无差异) | 10 | 2 | 20 |
| 4 | 扩增片段大于300 bp | 10 | 0 | 0 |
| 5 | 无扩增 | 10 | 0 | 0 |
2.5 豌豆品种指纹图谱构建
表5 9对SSR标记名称与信息
Table 5
| 引物名称Primer name | 上游引物Forward primer | 下游引物Reverse primer | 染色体Chromosome |
|---|---|---|---|
| PsSSR-6 | ACACAAATTCCCCACTCCCC | TCAACCGGTGGAAGAAACCC | Ps01 |
| PsSSR-27 | ACTTGTAAGCAGCTGAGCGT | GTAGCTGTGACTATCCCGGC | Ps02 |
| PsSSR-56 | TCCGGGCGAGACTACTTGAT | GTGGATTCCCATGGATCGCA | Ps04 |
| PsSSR-77 | TCCAGGCTTTGCATGATGGT | CCAACGACTCCAAGGCTTCA | Ps05 |
| PsSSR-80 | ATTTTGGCGGGGTTTTGACG | GTCCACCTCCGGGATCAAAG | Ps05 |
| PsSSR-81 | CCCCAATCACTTTCTCGTGC | GGAGCACACTCTCAAGACCC | Ps05 |
| PsSSR-82 | TGAAAGACAGGGATGCGCTT | AGGGCATGTACTTGCTTCGA | Ps05 |
| PsSSR-92 | TGGGTTCGTTCCCCTAATGG | CAGGTCAGGAGCAGAAGGTT | Ps06 |
| PsSSR-95 | CTGGGTGTGGTTCTTCACGA | TTGCTTACCTGCTTGCATGC | Ps06 |
表6 32份豌豆材料SSR标记指纹代码
Table 6
| 编号Code | 品种Variety | 指纹代码Fingerprint code | 编号Code | 品种Variety | 指纹代码Fingerprint code |
|---|---|---|---|---|---|
| 1 | DWD05-01 | 223142132 | 17 | 彩豌豆 | 313243233 |
| 2 | 成豌11号 | 423141432 | 18 | S5008 | 223241233 |
| 3 | C13 | 323142332 | 19 | RC09 | 222243233 |
| 4 | 方绿豌豆 | 323241132 | 20 | 食荚型甜豌豆 | 222243531 |
| 5 | 杂粮甜豌豆 | 302142532 | 21 | 鲜嫩二号豌豆 | 312343141 |
| 6 | WD-0030 | 432233132 | 22 | 美国甜脆豌豆 | 121143341 |
| 7 | 定豌7号 | 432123441 | 23 | 豌豆 | 223344313 |
| 8 | WD-0007 | 332213132 | 24 | 德利荷兰豆 | 221143443 |
| 9 | WD-0013 | 122121112 | 25 | WD-0019 | 121143343 |
| 10 | 麻豌豆 | 222223112 | 26 | 优选法国008香豆 | 322143344 |
| 11 | 澳洲红玉豌豆 | 421323431 | 27 | DWD05-03 | 133243444 |
| 12 | S6071 | 221322112 | 28 | 猪耳朵 | 222252444 |
| 13 | 黑眼豌豆 | 323321132 | 29 | WD-0009 | 321253434 |
| 14 | SW1102 | 223133122 | 30 | DWD05-010 | 221252444 |
| 15 | C01 | 323131233 | 31 | WD04-09 | 121143443 |
| 16 | 1702 | 313133133 | 32 | 黑豌豆 | 121243544 |
3 讨论
传统豌豆品种鉴定一般有幼苗形态鉴定[25]和形态标记鉴定[26],通过幼苗鉴定其生长容易受到环境因素的影响干扰鉴定结果;形态标记鉴定周期长且很多品种间性状差异小[27],使得品种鉴定非常困难。SSR分子标记具有共显性、单碱基高分辨率、操作简单、重复性好、可检测等位基因和随机均匀分布在扩增基因组内等优点[16],已在植物遗传多样性研究中得到广泛应用[13,28]。本研究利用豌豆v1a基因组,在豌豆7条染色体中鉴定出311 601个SSR位点,发现二碱基重复单元是最常见的类型,三碱基重复次之,四碱基重复最少,这与烟草[29]、拟南芥[30]、小麦[31]、西红柿和马铃薯[32]基因组SSR位点中含有大量二碱基和三碱基重复一致。豌豆SSR位点中,主要以A、T碱基组合为主,在二碱基重复单元中AT/TA(占30.76%)和TA/AT(占29.42%)占比最高,GC和 CG/GC占比不足1.00%,这与烟草四倍体野花相似。童治军等[29]发现,烟草中以二碱基重复为主,其中二碱基中AT/TA和CG/GC是出现最多和最少的基序结构。王玉龙[33]等发现四倍体野生种花生基因组中以A/T重复单元数量最多,其次是AT/AT、AAT/ATT,分别占基因组SSR的45.36%、19.26%、8.81%。
海量的基因组数据为定位SSR位点提供了便利,生物信息学分析方法为开发SSR分子标记提供了有效的工具[14]。但选择变多的同时也导致难以区分SSR分子标记的好坏,需要设计大量SSR分子标记进行筛选,这无疑增加了试验成本与时间。本研究结果表明,e-PCR方法可有效预判分子标记扩增结果,可以极大地提高豌豆基因组中多态性SSR标记的筛选效率,减少了人力和资金成本。
4 结论
在豌豆中运用e-PCR与真实PCR相结合的方法,利用e-PCR分析了122对SSR引物在4个豌豆基因组之间的特异性和多态性,发现46对具有多态性的SSR引物,真实PCR检测结果显示9对符合期望,能够使用,预测不具有多态性的SSR引物在真实PCR中仅有2对具有多态性。
参考文献
SSR标记技术及其在植物遗传学中的应用
Survey of plant short tandem DNA repeats
DOI:10.1007/BF00222386
PMID:24185874
[本文引用: 2]

Length variations in simple sequence tandem repeats are being given increased attention in plant genetics. Some short tandem repeats (STRs) from a few plant species, mainly those at the dinucleotide level, have been demonstrated to show polymorphisms and Mendelian inheritance. In the study reported here a search for all of the possible STRs ranging from mononucleotide up to tetranucleotide repeats was carried out on EMBL and GenBank DNA sequence databases of 3026 kb nuclear DNA and 1268 kb organelle DNA in 54 and 28 plant species (plus algae), respectively. An extreme rareness of STRs (4 STRs in 1268 kb DNA) was detected in organelle compared with nuclear DNA sequences. In nuclear DNA sequences, (AT)n sequences were the most abundant followed by (A)n · (T)n, (AG)n · (CT)n, (AAT)n · (ATT)n, (AAC)n · (GTT), (AGC)n · (GCT)n, (AAG)n · (CTT)n, (AATT)n · (TTAA)n, (AAAT)n · (ATTT)n and (AC)n · (GT)n sequences. A total of 130 STRs were found, including 49 (AT)n sequences in 31 species, giving an average of 1 STR every 23.3 kb and 1 (AT)n STR every 62 kb. An abundance comparable to that for the dinucleotide repeat was observed for the tri- and tetranucleotide repeats together. On average, there was 1 STR every 64.6 kb DNA in monocotyledons versus 1 every 21.2 kb DNA in dicotyledons. The fraction of STRs that contained G-C basepairs increased as the G+C contents went up from dicotyledons, monocotyledons to algae. While STRs of mono-, di- and tetranucleotide repeats were all located in non coding regions, 57% of the trinucleotide STRs containing G-C basepairs resided in coding regions.
Mutational dynamics of microsatellites
DOI:10.1007/s12033-009-9230-4
PMID:20012711
[本文引用: 1]
Microsatellites are a ubiquitous class of simple repetitive DNA sequences, which are widespread in both eukaryotic and prokaryotic genomes. The use of microsatellites as polymorphic DNA markers has considerably increased both in the number of studies and in the number of organisms, primarily for genetic mapping, studying genomic instability in cancer, population genetics, forensics, conservation biology, molecular anthropology and in the studies of human evolutionary history. Although simple sequence repeats have been extensively used in studies encompassing varied areas of genetics, the mutation dynamics of these genome regions is still not well understood. The present review focuses on the mutational dynamics of microsatellite DNA with special reference to mutational mechanisms and their role in microsatellite evolution.
Genome-wide microsatellite characteristics of five human Plasmodium species, focusing on Plasmodium malariae and P.ovale curtisi
DOI:10.1051/parasite/2020034
PMID:32410726
Microsatellites can be utilized to explore genotypes, population structure, and other genomic features of eukaryotes. Systematic characterization of microsatellites has not been a focus for several species of Plasmodium, including P. malariae and P. ovale, as the majority of malaria elimination programs are focused on P. falciparum and to a lesser extent P. vivax. Here, five human malaria species (P. falciparum, P. vivax, P. malariae, P. ovale curtisi, and P. knowlesi) were investigated with the aim of conducting in-depth categorization of microsatellites for P. malariae and P. ovale curtisi. Investigation of reference genomes for microsatellites with unit motifs of 1-10 base pairs indicates high diversity among the five Plasmodium species. Plasmodium malariae, with the largest genome size, displays the second highest microsatellite density (1421 No./Mbp; 5% coverage) next to P. falciparum (3634 No./Mbp; 12% coverage). The lowest microsatellite density was observed in P. vivax (773 No./Mbp; 2% coverage). A, AT, and AAT are the most commonly repeated motifs in the Plasmodium species. For P. malariae and P. ovale curtisi, microsatellite-related sequences are observed in approximately 18-29% of coding sequences (CDS). Lysine, asparagine, and glutamic acids are most frequently coded by microsatellite-related CDS. The majority of these CDS could be related to the gene ontology terms "cell parts," "binding," "developmental processes," and "metabolic processes." The present study provides a comprehensive overview of microsatellite distribution and can assist in the planning and development of potentially useful genetic tools for further investigation of P. malariae and P. ovale curtisi epidemiology.© V.B. Mathema et al., published by EDP Sciences, 2020.
Genome-wide microsatellite markers in castor (Ricinus communis L.): identification, development, characterization, and transferability in Euphorbiaceae
DOI:10.1016/j.indcrop.2020.112461 URL
Genome-wide characterization and analysis of microsatellite sequences in camelid species
DOI:10.1007/s13364-019-00458-x
[本文引用: 1]
Microsatellites or simple sequence repeats (SSRs) are among the genetic markers most widely utilized in research. This includes applications in numerous fields such as genetic conservation, paternity testing, and molecular breeding. Though ordered draft genome assemblies of camels have been announced, including for the Arabian camel, systemic analysis of camel SSRs is still limited. The identification and development of informative and robust molecular SSR markers are essential for marker assisted breeding programs and paternity testing. Here we searched and compared perfect SSRs with 1–6 bp nucleotide motifs to characterize microsatellites for draft genome sequences of the Camelidae. We analyzed and compared the occurrence, relative abundance, relative density, and guanine-cytosine (GC) content in four taxonomically different camelid species:Camelus dromedarius,C. bactrianus,C. ferus, andVicugna pacos. A total of 546762, 544494, 547974, and 437815 SSRs were mined, respectively. Mononucleotide SSRs were the most frequent in the four genomes, followed in descending order by di-, tetra-, tri-, penta-, and hexanucleotide SSRs. GC content was highest in dinucleotide SSRs and lowest in mononucleotide SSRs. Our results provide further evidence that SSRs are more abundant in noncoding regions than in coding regions. Similar distributions of microsatellites were found in all four species, which indicates that the pattern of microsatellites is conserved in family Camelidae.
The development and use of microsatellite markers for genetic analysis and plant breeding with emphasis on bread wheat
DOI:10.1023/A:1003910819967 [本文引用: 2]
Complete chloroplast genome of the inverted repeat-lacking species Vicia bungei and development of polymorphic simple sequence repeat markers
箭筈豌豆品种间遗传差异的SSR分析及指纹图谱构建
DOI:10.11686/cyxb2018226
[本文引用: 2]
转录因子(transcription factors,TFs)可以通过调控基因表达来调节多种生物学过程;目前随着基因组学大数据的不断开发,使得大量TFs基因家族得以鉴定,为TF-SSR标记的开发利用提供了宝贵资源。本研究基于已有的转录组数据,鉴定出箭筈豌豆转录因子282个;并从中设计了208对SSR引物,筛选出35对多态性高、稳定性好和谱带清晰的引物,进一步在30个箭筈豌豆品种中共扩增到288个等位基因;平均每个标记可产生8.23个等位基因,多态性比率(PPB)均为100%,多态信息含量(PIC)范围为0.42~1.00,平均值为0.80。另外,从35对引物中筛选出具备多个品种特征谱带的核心引物6对,可将供试的30个箭筈豌豆品种完全分开。同时,基于6对核心引物构建了箭筈豌豆品种测试的指纹图谱,可为箭筈豌豆DUS(特异性、一致性、稳定性)测试、新品种保护,以及遗传背景分析提供技术依据。
甘蓝全基因组锚定SSR标记分析
一种改良的植物DNA提取方法
DOI:10.3724/SP.J.1259.2013.00072
[本文引用: 1]
植物组织中含有大量多糖、多酚、酯类等次生代谢产物, 要从中提取高质量的DNA比较困难。针对这一情况, 该文提出一种改良CTAB植物DNA提取方法(mCTAB), 并以10种常见植物为实验材料, 与4种常用的植物DNA提取试剂盒作对比。结果表明, mCTAB法提取的DNA产率高且质量好, PCR扩增成功率也较高, 而提取成本显著低于DNA提取试剂盒, 可有效用于植物DNA条形码等研究的植物DNA提取。
普通烟草及其祖先种基因组SSR位点分析
DOI:10.3864/j.issn.0578-1752.2015.11.003
[本文引用: 2]
【目的】普通烟草(N. tabacum ,2n=24Ⅱ=48 TTSS)及其2个祖先种-绒毛状烟草(N. tomentosoformis,2n=12Ⅱ=24 TT)和林烟草(N.sylvestris,2n=12Ⅱ=24 SS)基因组SSR位点信息的统计分析,有助于烟草属植物的遗传分析。【方法】从公共数据库NCBI(National Center for Biotechnology Information)中下载上述3个烟草基因组数据,应用SSRIT和TRF软件分析其各自SSR位点分布特征,每个基因组随机合成50对SSR引物扩增多态性。【结果】在绒毛状烟草基因组、林烟草基因组和普通烟草基因组中分别获得218 081、263 478和397 432个SSR总位点,其间的平均距离分别为7.52、7.78和9.06 kb。绝大部分的SSR位点分布在内含子和UTR(尤其是5′-UTR)区域;以2核苷酸和3核苷酸类型为主,占基因组内SSR位点总数目的2/3以上,其中,2核苷酸类型丰度最高;含有A(T)<sub>n</sub>基序结构的频率及数量最高;除单核苷酸类型外,重复次数多在3—10。150对合成的引物对8个烟草种DNAs进行PCR反应,所有材料均能扩增出清晰稳定的目标片段,其中36对引物显示多态性。【结论】绒毛状烟草、林烟草和普通烟草基因组内SSR呈现一定的分布特征,表明SSR位点在亲缘关系相对较近的烟草种间具有高度保守性。
Distinct patterns of SSR distribution in the Arabidopsis thaliana and rice genomes
Data mining for simple sequence repeats in expressed sequence tags from barley, maize, rice, sorghum and wheat
DOI:10.1023/A:1014875206165 [本文引用: 1]
Large-scale identification of polymorphic microsatellites using an in silico approach
DOI:10.1186/1471-2105-9-374 [本文引用: 1]
四倍体野生种花生A.monticola全基因组SSR的开发与特征分析
DOI:10.3864/j.issn.0578-1752.2019.15.002
[本文引用: 1]
【目的】通过对四倍体野生种花生Arachis monticola(AABB,2n = 4x = 40)全基因组SSR位点搜索,研究其全基因组SSR分布特征及规律,开发并验证其全基因组SSR引物,为花生属植物遗传进化分析及重要性状分子标记开发提供依据。【方法】在华大基因GigaScience数据库下载A.monticola全基因组序列,并利用生物信息学软件MISA进行SSR位点搜索,Primer 3进行引物设计,通过电子PCR进行单位点SSR分析,并随机合成100对SSR引物验证通用性。【结果】SSR在四倍体野生种花生A.monticola基因组上共搜索到SSR位点676 878个,平均每3.8 kb就会出现一个SSR,分布于5 127条scaffold中,单核苷酸至六核苷酸均有分布,且数量上差异较大,以单碱基、二碱基、三碱基为主,三者占SSR总数的94.28%,其中单碱基重复数量最多,占46.71%,密度最高;六核苷酸重复数目最少,分布最稀疏。大多数SSR分布在基因间区,基因区SSR多分布于内含子区域;全基因组共鉴定出395个不同的重复基元,其中A亚基因组342种,B亚基因组356种;A/T是最丰富的重复基元;在1—6个核苷酸的重复基元中,数量最多的依次是A/T、AT/AT、AAT/ATT,AAAT/ATTT、AAAAT/ATTTT、AAAAAT/ATTTTT;整体来看,重复基元的重复次数多集中在50次以内,不同类型的motif的重复次数差异很大;同一种类型重复基元的SSR位点,随着motif重复次数增加,SSR的数量逐渐降低;B03染色体上SSR数量最多,A08染色体中SSR密度最高。A.monticola全基因组SSR比A.duranensis、A.ipaensis基因组SSR数量多,密度也更高,A.monticola单核苷酸重复最丰富,2个野生种二核苷酸数量最多。共设计出SSR引物192 303对,单位点SSR标记检出率50.35%,单点SSR标记在基因组上的分布呈现两端密集,中间稀疏的特点;随机合成的100对引物中,90对能在A. monticola中扩增出稳定清晰的条带,且在4份不同的花生基因组DNA中扩增目的条带表现出不同的特点。【结论】A.monticola基因组内SSR种类和数量丰富,单核苷酸至六核苷酸均有分布,单核苷酸重复基元数量最多,且最密,六核苷酸重复基元数量最少,出现频率最低,不同重复基元频数高低与核苷酸数量没有严格相关性,SSR多分布在基因间区,基因区内含子区域SSR数量最多;A.monticola A、B亚基因组具有其各自特异的重复基元类型;单个类型重复基元数量最多的均为AT富集的重复基元,而GC富集的重复基元相对较少;同一种类型重复基元的SSR位点,随着motif重复次数增加,SSR的数量逐渐降低;A.monticola全基因组SSR较2个二倍体野生种数量更多,密度也更高且重复基元分布规律不同;经过初步验证,开发的SSR引物在4份花生材料中表现出部分通用性。

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}