


开放科学(资源服务)标识码(OSID): 
云南是野生稻资源的重要富集区域,拥有元江、景洪、勐海、耿马和孟定等原生境,是中国及周边国家野生稻分布地域的水系源头,分布有我国全部3种野生稻:普通野生稻(AA基因组)、药用野生稻(Oryza Officinalis,CC基因组)和疣粒野生稻(GG基因组)。高原的立体性气候造就了云南野生稻丰富的生态类群,云南野生稻具有许多优异性状[1],其中药用野生稻有很强的抗病虫能力,高抗褐飞虱和白背飞虱,对叶蝉、螟虫和稻蓟马等也有很好的抗性;对白叶枯病和稻瘟病等有强的抗性。此外,药用野生稻生物量积累大,米粒中的蛋白质含量在12%以上,是对栽培稻遗传改良的重要资源[2,3,4,5,6]。药用野生稻与栽培稻基因组类型差异大,通过传统的有性杂交方式转移药用野生稻的优良性状和基因存在杂交不亲和、杂交不育等生殖障碍及重组频率低、不易存活等问题[7,8,9]。因此,构建基因组文库,建立药用野生稻渗入系库,是研究和发掘利用药用野生稻优良基因的途径[10]。
1 材料与方法
1.1 试验材料
药用野生稻材料为耿马居群,种植在云南省农业科学院生物技术与种质资源研究所的温室内。BIBAC载体购自美国康奈尔大学。
1.2 试验方法
1.2.1 制备核悬液 取50g健康的药用野生稻嫩叶,用灭菌蒸馏水漂洗2遍,于液氮中研磨成粉末,转移到500mL冰浴的溶液Ⅰ(含10mmol/L Tris碱,10mmol/L EDTA,1mmol/L亚精胺三盐酸盐,1mmol/L亚精胺三氨四盐酸盐,0.5mol/L蔗糖,0.5% Triton X-100,0.15%巯基乙醇)重悬,用磁力搅拌器轻漩涡10min,重悬液用3层神奇滤布(Miracloth,美国Millipore公司产品)过滤,挤压收集滤液,即为核悬液。
1.2.2 分离细胞核 将核悬液分装于50mL离心管,3 800g、4℃离心20min,弃上清,沉淀即为细胞核,用40mL冰冷的溶液Ⅰ重悬,经2层神奇滤布,通过静止过滤,滤液离心(3 800g、4℃、20min),再用溶液Ⅰ重悬洗涤1次,离心(3 800g、4℃、15min),沉淀即为细胞核。
1.2.3 细胞核包埋 细胞核重悬于1mL溶液Ⅱ(含10mmol/L Tris碱、10mmol/L EDTA、1mmol/L亚精胺三盐酸盐、1mmol/L亚精氨四盐酸盐和0.5mol/L蔗糖),用溶液Ⅱ配制2%的低熔点琼脂糖,将核悬液和低熔点琼脂糖于45℃中水浴,以1:1的比例混合后倒入冰上预冷的2mL医用注射器中,冷却后形成核包埋块。
1.2.4 细胞核裂解 依据注射器的刻度,推出凝固的琼脂包埋块,将其切成100µL的核包埋块,放入30mL裂解液(0.5mol/L pH=9.0 EDTA,1%月桂酰基肌氨酸钠和1mg/mL蛋白酶K)中,于恒温摇床(50℃、70转/min)中轻摇24h,将裂解完成后的包埋块置于冰浴中,于摇床(室温、70转/min)上清洗,各清洗液需预冷。清洗步骤:用60mL的50mmol/L EDTA(pH=8.0)清洗1次,每次1h,用60mL的TE缓冲液清洗1次,每次1h,用60mL含0.1mmol/LPMSF的TE缓冲液清洗3次,每次3h,用60mL的TE缓冲液清洗3次,每次3h。清洗后的核包埋块在TE中于4℃保存。
1.2.5 酶解 对酶用量和酶解时间进行优化。将每个核包埋块切成相连接的12片,用酶解缓冲液(灭菌双蒸水797µL、10×BamHⅠreaction buffer 100µL、1mol/L亚精胺2µL和1mol/L二硫苏糖醇1µL)于冰上预平衡1h,中途置换1次酶解缓冲液。平衡结束后加入酶解反应液(灭菌双蒸水72.5µL、10×BamHⅠreaction buffer 17µL、1mol/L亚精胺0.34µL、1mol/L二硫苏糖醇0.17µL、10mg/mL BSA 10µL和BamHⅠ0.2~1.4U),于冰上保存90min,然后转入37℃水浴中酶解3~8min,加入20µL预冷的0.5mol/L EDTA(pH=8.0)于冰上终止反应。
1.2.6 电泳检测 经酶解的核包埋块、对照核包埋块和酵母PFG Marker凝胶块贴于制胶梳齿上,倒入0.5% TBE缓冲液配制的1%琼脂糖(PFGE级)凝胶,冷却后进行脉冲电泳(auto algorithm mode:225kb~1.9Mb,6V/cm,120°,linear,16h,其余参数为默认值),电泳结束后于0.5µg/mL溴化乙锭中染色30min,于凝胶成像系统中检测拍照。
1.2.7 目标DNA的收集 用冰冷的灭菌去离子水清洗透析袋数次,再用冰冷的0.5% TBE缓冲液冲洗数次,从凝胶上选择DNA目标片段区域(未经紫外线照射),切胶放入透析袋,加入冰冷的0.5% TBE缓冲液,排尽空气,两端用透析袋夹子密封,置于脉冲场中电泳(two state,6V/cm,120°,35s,35s,linear,5h,其余参数为默认值),透析袋与电场方向垂直。电泳结束后,将透析袋倒转180°放置,继续电泳1min,回收透析袋中的溶液,放入新的透析袋中,于冰冷的灭菌去离子水中透析,去除DNA溶液中的离子,经透析后的DNA即用于后续的载体连接。
2 结果与分析
2.1 药用野生稻叶片细胞核的分离
药用野生稻植株高大,叶片宽且长,为了分离完整和数量足够的叶片细胞核,应选取药用野生稻嫩叶,加液氮研磨成粉末,且研磨要适度。将所取的药用野生稻嫩叶不剪断,轻卷后全部浸于液氮中,先用钵棒捣碎,再进行研磨。可用一大一小2个研钵研磨,将大研钵中研磨产生的上层组织块用镊子挑到小研钵中继续研磨,既可研磨充分,又避免过度研磨。未完成研磨之前应尽可能将材料浸于液氮中,防止反复冻融损伤DNA。用3层滤布和2层滤布各静止过滤1次,去除组织杂质;选择3 800g离心力,既能满足细胞核沉降完全,又能减少离心力的破坏。经过过滤、离心和洗涤,分离出数量可观、完整、分散好的药用野生稻叶片细胞核(图1)。
图1

图1
药用野生稻叶片细胞核分离
A.药用野生稻;B.叶片匀浆液;C.细胞核离心;D.细胞核离心和清洗;E.细胞核
Fig.1
Nucleus separation from leaves of O.officinalis
A. O.officinalis; B. Leaves homogenate; C. Nuclear centrifugation; D. Nuclear centrifugation and cleaning; E. Nucleus
2.2 药用野生稻叶片细胞核的包埋与洗涤
为了将细胞核中的染色体DNA裂解释放,需要进行核包埋。1%的低熔点琼脂糖可产生软硬合适的固块,同时保证合适的孔隙以利于裂解液的浸入和DNA的释放;利用自制的医用注射器进行核包埋,有利于控制切片厚度和大小。裂解和洗涤均于摇床中进行,用冰浴进行控温,保证裂解充分。经过核包埋、切块、裂解和洗涤,得到裂解后的细胞核(图2)。
图2
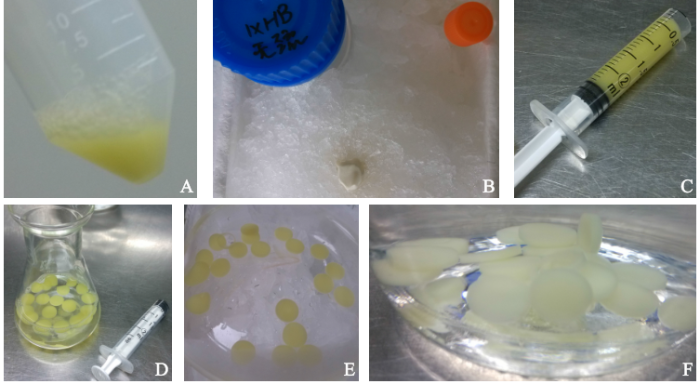
图2
药用野生稻叶片细胞核的低熔点琼脂糖包埋
A.核重悬;B.琼脂糖在冰上凝固;C.包埋块凝固于医用注射器中;D.包埋块切成圆片;E.嵌入式块清洗;F.包埋块洗涤
Fig.2
Nucleus embedding of O. officinalis by low-melting point agarose
A. Nuclear resuspension; B. Agarose solidified on the ice; C. Embedding block is solidified in the medical syringe; D. Cut into a circular piece; E. Embedded block washing; F. Embedded block after washing
2.3 药用野生稻染色体DNA的释放与消化
为了分离出完整的染色体DNA,得到合适大小的用于建库的DNA片段,对裂解后的细胞核进行部分酶解。结果表明,未经酶解的染色体DNA带型较为完整,几乎无降解,使用BamH I酶解后,DNA被切割成不同大小的片段。根据目标片段大小要求,分析不同酶解时间及酶量对DNA酶解的影响,发现酶解7min时,最佳的酶用量为0.6U(图3);0.6U酶量下,最适酶解时间为7min(图4)。在这2个最佳条件下,染色体DNA酶解彻底,DNA片段集中度好(图5)。用于构建BIBAC文库的质粒为23.5kb,它所能装载的最大片段可达150kb(图6)。通过本提取方法,从药用野生稻中成功分离出高质量的染色体DNA,片段大小达1.9Mb,DNA经酶解、电洗脱和透析纯化回收,经电泳检测和微量核酸浓度测定仪检测,回收DNA最大浓度为90ng/µL(图5和图6),可用于基因组文库构建、基因及基因组保存、基因鉴定。
图3

图3
不同浓度BamH I处理8min对药用野生稻基因组DNA消化的影响
M.酵母染色体PFG marker;Non.未经BamH I消化。下同
Fig.3
Effects of different concentrations of BamH I on genomic DNA digestion of O.officinalis under eight minutes treatment
M. Yeast chromosome PFG marker; Non. DNA without enzyme treatment. The same below
图4
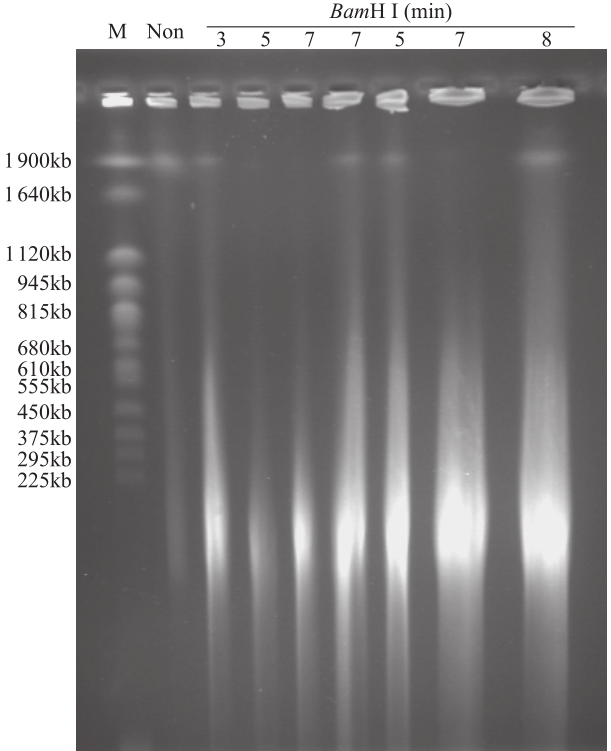
图4
0.6U BamH I处理不同时间对药用野生稻基因组DNA消化的影响
Fig.4
Effects of digestion time on genomic DNA of O. officinalis under 0.6U BamH I treatment
图5
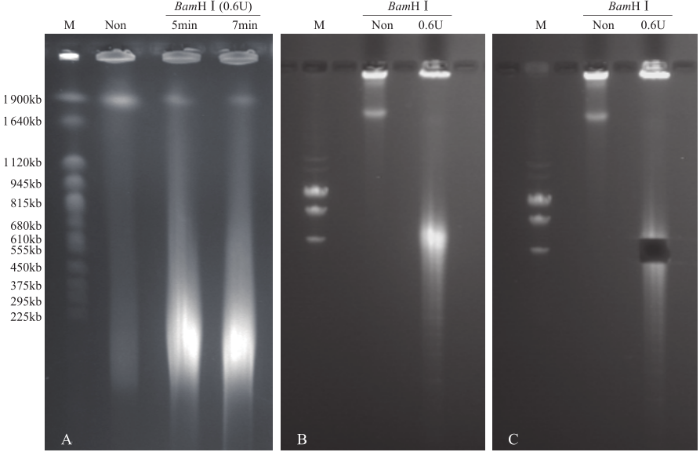
图5
药用野生稻染色体DNA未经及经BamHⅠ部分消化
A.最适酶解条件,B和C.酶解消化后目标DNA区域的切取
Fig.5
Chromosome DNA of O. officinalis without or with BamHⅠdigestion
A. Optimum conditions for BamHⅠ, B and C. Extraction of target DNA region after enzymatic digestion
图6
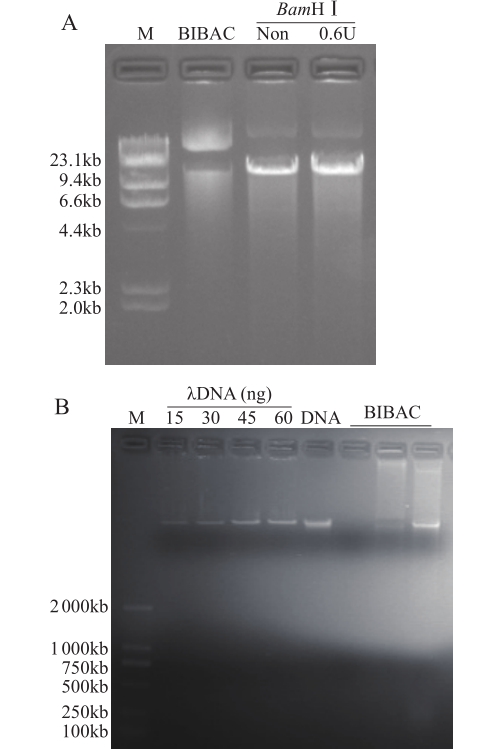
图6
BIBAC质粒及药用野生稻染色体大片段回收
A. BIBAC质粒及酶解,B.染色体大片段回收;M. λDNA/HindⅢ marker (A)和D2000 marker (B)
Fig.6
BIBAC vector and large-fragment DNA recovery of O.officinalis
A. BIBAC and digested by BamHⅠ, B. Target DNA recovery for BamHⅠ;M. Yeast chromosome PFG marker (A) and D2000 marker (B)
3 讨论
药用野生稻是我国现有的3种野生稻之一,具有抗病虫害、抗逆境胁迫、高光效和高产优质等优良特性,是宝贵的基因库[19,20]。药用野生稻耐贫瘠,吸收矿质元素能力强,根茎叶的组织解剖结构与C4植物有一定相似性,如维管束发达[21,22]。最值得关注的是,药用野生稻生长势强,生物量积累大(约为栽培稻的20倍以上),在温室株高可超过2m,在野外原生境可高达4m[1],这可能与其光合效率有关,药用野生稻中确实含有高光效基因(结果未发表)。药用野生稻远缘杂交不亲合,难以获得后代材料并迅速扩大群体。本研究创制的药用野生稻杂交后代群体回交到BC4F1仍不结实,因此,优化提取体系和流程,获得高质量的药用野生稻染色体DNA是对其优良性状和所蕴含的优异基因研究利用的前提,对构建大片段基因组文库有重要意义。BIBAC基因组文库技术能实现基因成簇转移[23,24,25],减少基因沉默及位置效应,更多地保持目的性状代谢网络中的酶及调控因子,从而实现更大限度的遗传改良。国内外学者利用该技术建立了蕃茄[25]、拟南芥[26]、盐芥[27]、小桐子[28]及棉花[29]等植物的BIBAC文库,包括东乡野生稻[30]和药用野生稻[17-18,31]。
与已报道的方法[17,31]相比,本研究在4个方面进行了改进:一是取样部位和取样量,本试验所取的是刚抽展出的药用野生稻嫩叶,并拨出隐藏在叶原基以上未见光的黄化叶部分,以降低叶片中色素等次生代谢产物的干扰,且本研究取样量大(50g),可得到数量可观的细胞核。二是过滤和离心参数,传统方法使用16层纱布、250目筛网过滤,洗涤离心5次,离心力1 800g;本研究参数为:重悬转数300转/min,3层神奇滤布过滤1次,离心1次重悬,2层滤布再过滤1次,沉淀洗涤1次,离心力3 800g,减少了样品吸附消耗量,去杂效果更好,减小了剪切力对细胞核的伤害,时间也由原来的2h缩短到1h。三是包埋方法,传统方法是45℃,1.5%低融点琼脂糖,使用凝胶块模子;本研究设定40℃,2%低融点琼脂糖,使用2mL医用注射器。相比之下,降低5℃有利于减少DNA的高温降解,使用2mL医用注射器可加快包埋速度,可依据刻度控制切割成任意大小的核包埋块。四是酶解方法,传统方法是:0.4mg/mL蛋白酶K先解离,再用BamHⅠ部分酶解。本试验使用0.1%蛋白酶K(相当于1mg/mL),BamHⅠ酶解条件为0.6U、37℃、7min。在这一条件下,染色体组蛋白解离充分,DNA释放完全,不会产生蛋白污染。
4 结论
在前人染色体DNA提取方法的基础上,从取材与研磨、细胞核的分离与包埋、组蛋白的去除与DNA释放和DNA的酶解与回收等环节对药用野生稻染色体DNA的提取方法进行了优化,最终成功提取出1.9Mb大小的染色体DNA,并优化了酶切消化体系。
参考文献
药用野生稻有利基因发掘与利用研究进展
药用野生稻具有丰富的遗传多样性并含有大量优异基因。对药用野生稻的利用主要存在遗传学背景不清楚、常规育种效率不高两方面的障碍。这些问题可以通过构建药用野生稻可转化大片段基因组文库和转基因技术,将大片段克隆导入栽培稻中,建立全基因组基因嵌入突变体库来解决。本文对国内外药用野生稻有利基因发掘及其利用的研究进展进行综述。
云南三种野生稻原生境植物种群的调查及比较分析
对云南省境内的3种野生稻40个原生境的植物群落进行了考察,共记录植物159种,分属于51科。按乔木层、灌木层、草本层对野生稻原生境植物种类进行分类,乔木层植物共6科1亚科11种,灌木层植物共29科60种,草本层植物共20科88种;普通野生稻原生境的植物种类95%为草本层,疣粒野生稻的植物则有明显的垂直层次结构。普通野生稻原生境植物共计20种,均为特有植物种,分属于5科,主要为禾本科和莎草科;药用野生稻原生境植物共计25种,分属于19个科,特有种15个;疣粒野生稻原生境植物共计125种,分属于45科,特有植物种数达117个。最后就3种野生稻原生境植物种类调查的意义、外来物种对野生稻的原生境威胁等问题进行了讨论。
茶陵普通野生稻光合特性研究
以超高产或高产栽培稻两优培九、汕优63、威优46和93-11为对照,测定了湖南茶陵普通野生稻抽穗开花期1 d中不同时间点的净光合速率(Pn)以及Pn对不同光强、不同CO2浓度与不同温度的响应曲线。结果表明,茶陵普通野生稻的Pn在下午极显著高于对照,在高温(40℃,45℃)胁迫下也是如此;光补偿点为22.3 µmol photons m-2 s-1,与对照差异不显著(20.1~23.7 µmol photons m-2 s-1),光饱和点为1 810 µmol photons m-2 s-1,与对照93-11无显著差异,但极显著高于其他对照材料(1 530~1 628 µmol photons m-2 s-1),表观量子效率(AQY)与对照无显著差异;CO2补偿点(52.7 μmol mol-1)稍高于对照(42.7~50.1 μmol mol-1),而饱和点(644.5 μmol mol-1)则明显高于对照(521.1~581.3 μmol mol-1),羧化效率(0.1511 mol m-2 s-1)显著高于对照(0.1277~0.1384 mol m-2 s-1);叶绿素含量极显著高于对照。说明茶陵普通野生稻的光合性能强于超高产或高产对照栽培稻,且在高温下表现更为突出。]]>
Stable transfer of intact high molecular weight DNA into plant chromosomes
A binary-BAC system for plant transformation with high-molecular-weight DNA
DOI:10.1016/S0378-1119(97)00388-0 URL [本文引用: 1]
Construction of tomato genomic DNA libraries in a binary-BAC (BIBAC) vector
DOI:10.1046/j.1365-313X.1999.00433.x URL [本文引用: 2]
Characterization of a plant-transformation-ready large-insert BIBAC library of Arabidopsis and bombardment transformation of a large-insert BIBAC of the library into tobacco
DOI:10.1139/G11-011
URL
[本文引用: 1]

Plant-transformation-ready, large-insert binary bacterial artificial chromosome (BIBAC) libraries are of significance for functional and network analysis of large genomic regions, gene clusters, large-spanning genes, and complex loci in the post-genome era. Here, we report the characterization of a plant-transformation-ready BIBAC library of the sequenced Arabidopsis genome for which such a library is not available to the public, the transformation of a large-insert BIBAC of the library into tobacco by biolistic bombardment, and the expression analysis of its containing genes in transgenic plants. The BIBAC library was constructed from nuclear DNA partially digested with BamHI in the BIBAC vector pCLD04541. It contains 6144 clones and has a mean insert size of 108 kb, representing 5.2x equivalents of the Arabidopsis genome or a probability of greater than 99% of obtaining at least one positive clone from the library using a single-copy sequence as a probe. The transformation of the large-insert BIBAC and analyses of the transgenic plants showed that not only did transgenic plants have intact BIBAC DNA, but also could the BIBAC be transmitted stably into progenies and its containing genes be expressed actively. These results suggest that the large-insert BIBAC library, combined with the biolistic bombardment transformation method, could provide a useful tool for large-scale functional analysis of the Arabidopsis genome sequence and applications in plant-molecular breeding.
A large insert Thellungiella halophila BIBAC library for genomics and identification of stress tolerance genes
Construction and characterization of a BIBAC library of Jatropha curcas L.and identification of BIBAC clones containing genes associated with fatty acid metabolism
DOI:10.1007/s11032-010-9505-2
URL
[本文引用: 1]
Jatropha curcas L. is a potentially significant bioenergy crop in the tropics and subtropics. Here we present a plant-transformation-competent binary bacterial artificial chromosome (BIBAC) library from Jatropha cultivar YN049-4. This library was constructed with BamH in the vector pCLD04541, consists of 30,720 clones and is arrayed in 80 384-well microtiter plates. Since 92.1% (28,293) of its clones were shown to contain Jatropha DNA inserts with an average size of 131.9 kb, the library is estimated to represent approximately 8.9 haploid genome equivalents of the species, thus providing a greater than 99% probability of discovering a particular single-copy sequence in the library. High-density clone filters were made from a subset of the library and hybridized with nine pairs of overgos designed from genes involved in fatty acid metabolism. Hybridization results showed that eight overgo pairs were able to identify positive clones from the subset of the library, with an average of 5.3 clones per probe, suggesting that it is suitable for Jatropha genomics and genetics research. Because this library, to our knowledge, represents the first large-insert, plant-transformation-competent BIBAC library for Jatropha, it will provide a vital resource for advanced genomics research, including isolation and characterization of genes and quantitative trait loci, integrative physical mapping and genome sequencing.
Construction of a plant-transformation-competent BIBAC library and genome sequence analysis of polyploid upland cotton (Gossypium hirsutum L.)
DOI:10.1186/1471-2164-14-208
URL
PMID:23537070
[本文引用: 1]
BACKGROUND: Cotton, one of the world's leading crops, is important to the world's textile and energy industries, and is a model species for studies of plant polyploidization, cellulose biosynthesis and cell wall biogenesis. Here, we report the construction of a plant-transformation-competent binary bacterial artificial chromosome (BIBAC) library and comparative genome sequence analysis of polyploid Upland cotton (Gossypium hirsutum L.) with one of its diploid putative progenitor species, G. raimondii Ulbr. RESULTS: We constructed the cotton BIBAC library in a vector competent for high-molecular-weight DNA transformation in different plant species through either Agrobacterium or particle bombardment. The library contains 76,800 clones with an average insert size of 135 kb, providing an approximate 99% probability of obtaining at least one positive clone from the library using a single-copy probe. The quality and utility of the library were verified by identifying BIBACs containing genes important for fiber development, fiber cellulose biosynthesis, seed fatty acid metabolism, cotton-nematode interaction, and bacterial blight resistance. In order to gain an insight into the Upland cotton genome and its relationship with G. raimondii, we sequenced nearly 10,000 BIBAC ends (BESs) randomly selected from the library, generating approximately one BES for every 250 kb along the Upland cotton genome. The retroelement Gypsy/DIRS1 family predominates in the Upland cotton genome, accounting for over 77% of all transposable elements. From the BESs, we identified 1,269 simple sequence repeats (SSRs), of which 1,006 were new, thus providing additional markers for cotton genome research. Surprisingly, comparative sequence analysis showed that Upland cotton is much more diverged from G. raimondii at the genomic sequence level than expected. There seems to be no significant difference between the relationships of the Upland cotton D- and A-subgenomes with the G. raimondii genome, even though G. raimondii contains a D genome (D5). CONCLUSIONS: The library represents the first BIBAC library in cotton and related species, thus providing tools useful for integrative physical mapping, large-scale genome sequencing and large-scale functional analysis of the Upland cotton genome. Comparative sequence analysis provides insights into the Upland cotton genome, and a possible mechanism underlying the divergence and evolution of polyploid Upland cotton from its diploid putative progenitor species, G. raimondii.
Discrimination between Oryza malampuzhaensis Krish. et Chand. and Oryza officinalis Wall ex Watt based on RAPD markers and morphological traits
DOI:10.1023/A:1012602516236
URL
[本文引用: 1]
Oryza malampuzhaensis, one of the tetraploid taxa of the genusOryza, is geographically restricted to Western Ghats of South India. Its taxonomic status is not well established and is generally treated as a tetraploid race of O. officinalis. Sixty-three morphological traits and 262 random amplified polymorphic DNA (RAPD) markers generated by 23 random decamer primers were used to assess the genetic relationship between O. malampuzhaensis and O. officinalis. Pair wise comparisons based on both RAPDs and morphological traits revealed 60% genetic distance between the two taxa and was significantly higher (p <0.01) than the corresponding intra-specific distances. Cluster and principal component analysis (PCA) of genetic distance estimations based on RAPDs and morphological traits clearly differentiated the two taxa. High frequency of discrete O. malampuzhaensis specific RAPDs (21%) and the significantly higher (p < 0.05) mean number of amplification products per individual in O. malampuzhaensis observed in the study reflect its allopolyploid nature. Low genetic diversity within O. malampuzhaensis revealed by RAPD analysis indicates the recent origin of this taxa. The RAPD analysis further revealed the possibility that the putative ‘C’ genomeprogenitor of O. malampuzhaensis is a close relative of O. officinalis. In addition, amplification products diagnostic to O. malampuzhaensis were identified. The results of the present study clearly demonstrated that the O. malampuzhaensis is a distinct entity, and support the recent conclusion that O. malampuzhaensis has diverged enough to deserve species status.]]>
Genetic diversity patterns in ex situ collections of Oryza officinalis Wall. ex G. Watt revealed by morphological and microsatellite markers
DOI:10.1007/s10722-016-0396-x URL [本文引用: 1]
Multi-step formation, evolution, and functionalization of new cytoplasmic male sterility genes in the plant mitochondrial genomes
DOI:10.1038/cr.2016.115
URL
PMID:27725674
[本文引用: 1]
New gene origination is a major source of genomic innovations that confer phenotypic changes and biological diversity. Generation of new mitochondrial genes in plants may cause cytoplasmic male sterility (CMS), which can promote outcrossing and increase fitness. However, how mitochondrial genes originate and evolve in structure and function remains unclear. The rice Wild Abortive type of CMS is conferred by the mitochondrial gene WA352c (previously named WA352) and has been widely exploited in hybrid rice breeding. Here, we reconstruct the evolutionary trajectory of WA352c by the identification and analyses of 11 mitochondrial genomic recombinant structures related to WA352c in wild and cultivated rice. We deduce that these structures arose through multiple rearrangements among conserved mitochondrial sequences in the mitochondrial genome of the wild rice Oryza rufipogon, coupled with substoichiometric shifting and sequence variation. We identify two expressed but nonfunctional protogenes among these structures, and show that they could evolve into functional CMS genes via sequence variations that could relieve the self-inhibitory potential of the proteins. These sequence changes would endow the proteins the ability to interact with the nucleus-encoded mitochondrial protein COX11, resulting in premature programmed cell death in the anther tapetum and male sterility. Furthermore, we show that the sequences that encode the COX11-interaction domains in these WA352c-related genes have experienced purifying selection during evolution. We propose a model for the formation and evolution of new CMS genes via a
Genetic mechanisms of abiotic stress tolerance that translate to crop yield stability
DOI:10.1038/nrg3901
URL
PMID:25752530
[本文引用: 1]
Crop yield reduction as a consequence of increasingly severe climatic events threatens global food security. Genetic loci that ensure productivity in challenging environments exist within the germplasm of crops, their wild relatives and species that are adapted to extreme environments. Selective breeding for the combination of beneficial loci in germplasm has improved yields in diverse environments throughout the history of agriculture. An effective new paradigm is the targeted identification of specific genetic determinants of stress adaptation that have evolved in nature and their precise introgression into elite varieties. These loci are often associated with distinct regulation or function, duplication and/or neofunctionalization of genes that maintain plant homeostasis.
Multiple origins of BBCC allopolyploid species in the rice genus (Oryza)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}