


锌指蛋白是存在于真核生物中最大的转录因子家族之一,其三维结构包括2个平行的β折叠和1个与锌离子稳定结构结合的α螺旋,形似“手指”,因此得名。根据锌指基序类型的不同可以将锌指蛋白分为C2H2、C2HC、C2HC5、C2C2、CCCH、C3HC4、C4、C4HC3、C6和C8共10种类型(C代表半胱氨酸,H代表组氨酸)[1],其中C2H2型锌指蛋白是其中占比最大的一类。Han等[2]在矮牵牛花中发现了首个植物中的C2H2型锌指蛋白—EPF1,并且发现了14个与EPF1相关的基因。此后,陆续在拟南芥[3]、水稻[4]、番茄[5]、杨树[6]、大豆[7]、甘蓝[8]、木薯[9]和马铃薯[10]等多种植物中发现了C2H2型锌指蛋白,分别在植物生长发育、基因表达以及在应对生物及非生物胁迫中起到重要调控作用[11]。
甜菜(Beta vulgaris L.)是温带气候地区的一种重要作物,提供了世界上每年近30%的糖产量,同时也是新兴的可再生能源作物,乙醇转化率和生物学产量高,在生物质能开发利用方面具有潜在的用途[16]。同时,甜菜在重金属污染特别是镉污染的生物修复方面具有潜在的优势,主要体现在生长快速、根系能深植土壤、容易收割、能够容纳并累积多样化重金属等,并且甜菜的生长周期短,成活率高,生物量大,是适合于重金属污染修复的优良植物材料[17]。因此,挖掘镉胁迫下甜菜的应答基因对利用甜菜进行重金属污染生物修复和生物质能开发十分重要。目前,国内外关于甜菜锌指蛋白方面的研究鲜有报道,因此,本研究对甜菜中的C2H2锌指蛋白转录因子进行了全基因组鉴定,通过生物信息学手段对家族成员进行了分析,同时对镉胁迫下甜菜C2H2型锌指家族成员进行了表达分析和调控网络预测,旨在为甜菜C2H2型锌指蛋白的研究及耐镉甜菜种质的创制奠定基础。
1 材料与方法
1.1 植物材料处理及转录组测序
所用植物材料为黑龙江大学国家甜菜种质中期库提供的“780016B/12优”,试验在黑龙江大学国家甜菜种质中期库进行。选取大小均一的甜菜种子置于70%乙醇溶液中,震荡后用蒸馏水冲洗,在2‰福美双溶液中浸泡过夜,蒸馏水冲洗干净后将种子均匀铺撒在湿润的蛭石中并转移至人工气候室培养10 d,然后将甜菜苗转移至Hoagland营养液中水培。水培条件为白天25 ℃,晚上18 ℃,光照周期为14 h光照/10 h暗培养循环,光照强度为200 µmol/(m2·s),每3 d更换一次营养液。14 d后进行镉胁迫处理,选取长势均一的甜菜苗分别在0、1、3和5 mmol/L CdCl2胁迫下处理6 h,收集处理样本,测序工作由诺禾致源生物信息科技有限公司(北京,中国)进行。
1.2 甜菜C2H2型锌指蛋白家族的筛选与鉴定
从EnsemblPlants数据库(
1.3 甜菜C2H2型锌指蛋白家族进化树分析
对甜菜C2H2型锌指转录因子进行系统发育分析,用MEGA 7[19]的邻近法构建了系统发育进化树。从PlantTFDB(
1.4 染色体定位和基因结构分析
采用TBtools软件,从甜菜基因组注释信息中获取甜菜C2H2型锌指蛋白转录因子的染色体位置信息,对其进行可视化处理,将cDNA与基因组DNA序列进行比对,构建C2H2型锌指蛋白家族成员的基因结构。
1.5 理化特性分析、信号肽预测及亚细胞定位预测
应用ExPASy在线软件(
1.6 顺式作用元件分析
利用TBtools软件提取甜菜C2H2型锌指蛋白家族全部成员上游2000 bp的序列,将提取到的序列提交到PlantCARE(
1.7 镉胁迫下基因表达分析及转录因子调控网络分析
从转录组测序数据中提取家族成员在4个处理条件下的表达量(FPKM),利用微生信在线网站进行热图绘制。结合转录组测序结果筛选3个镉胁迫浓度下|log2(FoldChange)|>1且q值<0.005的差异表达基因,并对筛选结果取交集,利用TBtools提取差异表达基因的上游2000 bp序列提交到PlantTFDB进行转录因子结合位点预测,阈值设置为10-5。利用软件Gephi 0.10.1(
2 结果与分析
2.1 甜菜C2H2型锌指蛋白家族的筛选鉴定及分析
表1 甜菜C2H2型锌指蛋白家族成员理化性质及亚细胞定位
Table 1
| 基因ID Gene ID | 转录本ID Transcript ID | 氨基酸数量 Number of amino acids | 分子量 Molecular weight (u) | 等电点 Isoelectric point | 不稳定系数 Instability index | 亚细胞定位 Subcellular localization |
|---|---|---|---|---|---|---|
| BVRB_1g018520 | KMT00022 | 366 | 41 146.19 | 8.46 | 70.84 | 细胞核 |
| BVRB_6g136840 | KMT08971 | 534 | 59 773.12 | 9.10 | 59.57 | 细胞核 |
| BVRB_9g212520 | KMT01234 | 342 | 37 043.98 | 8.84 | 71.81 | 细胞外,细胞核 |
| BVRB_5g113850 | KMT10727 | 563 | 62 233.31 | 8.77 | 56.24 | 细胞核 |
| BVRB_1g012990 | KMT19293 | 456 | 50 064.47 | 5.93 | 60.88 | 细胞核 |
| BVRB_9g206210 | KMT02271 | 343 | 37 459.07 | 6.24 | 60.35 | 细胞核 |
| BVRB_7g179220 | KMS97061 | 340 | 37 135.81 | 7.75 | 55.44 | 细胞核 |
| BVRB_8g189640 | KMT03820 | 242 | 25 661.67 | 8.09 | 53.01 | 细胞核 |
| BVRB_1g015270 | KMT19130 | 275 | 29 083.48 | 8.78 | 60.67 | 细胞核 |
| BVRB_2g037310 | KMT17528 | 186 | 20 667.45 | 8.86 | 57.60 | 细胞核 |
| BVRB_1g018510 | KMT00021 | 523 | 56 286.76 | 6.37 | 51.56 | 细胞核 |
| BVRB_7g174490 | KMT05313 | 231 | 25 076.07 | 5.84 | 38.19 | 细胞核 |
| BVRB_2g046350 | KMS99266 | 543 | 60 544.44 | 7.17 | 39.85 | 细胞核 |
| BVRB_5g111450 | KMT11057 | 237 | 26 905.18 | 8.52 | 59.79 | 细胞核 |
| BVRB_6g147790 | KMT07654 | 226 | 25 663.52 | 9.10 | 62.68 | 细胞核 |
| BVRB_5g106070 | KMT11669 | 187 | 21 346.69 | 5.52 | 59.95 | 细胞核 |
| BVRB_6g130850 | KMT09633 | 208 | 22 645.27 | 8.60 | 47.07 | 细胞核 |
| BVRB_3g067630 | KMS98868 | 154 | 17 264.82 | 5.64 | 63.00 | 细胞核 |
| BVRB_3g067720 | KMS98878 | 247 | 27 624.67 | 9.20 | 45.64 | 细胞核 |
| BVRB_007850 | KMS95480 | 371 | 41 815.85 | 6.12 | 41.96 | 细胞核 |
| BVRB_3g068910 | KMS98817 | 309 | 34 263.02 | 8.70 | 58.91 | 细胞核 |
| BVRB_3g063640 | KMT15255 | 323 | 35 834.41 | 8.56 | 58.90 | 细胞核 |
| BVRB_6g138130 | KMT08530 | 1460 | 164 803.16 | 6.37 | 42.75 | 细胞核 |
| BVRB_016580 | KMS94655 | 176 | 19 758.12 | 6.15 | 48.27 | 细胞核 |
| BVRB_6g141700 | KMT08346 | 247 | 25 923.68 | 8.64 | 68.65 | 细胞核 |
| BVRB_007840 | KMS95479 | 414 | 46 178.78 | 8.58 | 53.94 | 细胞核 |
| BVRB_007870 | KMS95482 | 370 | 41 964.66 | 8.12 | 44.37 | 细胞核 |
| BVRB_007880 | KMS95483 | 406 | 45 560.31 | 7.52 | 46.30 | 细胞核 |
| BVRB_1g000840 | KMT20065 | 513 | 57 833.77 | 5.84 | 49.07 | 细胞核 |
| BVRB_6g137660 | KMT09067 | 559 | 58 440.17 | 8.62 | 72.22 | 细胞核 |
| BVRB_042870 | KMS64767 | 88 | 10 142.79 | 11.72 | 50.60 | 叶绿体,线粒体,细胞核 |
| BVRB_5g123490 | KMS97785 | 641 | 73 777.76 | 5.00 | 56.65 | 高尔基体,细胞核 |
图1
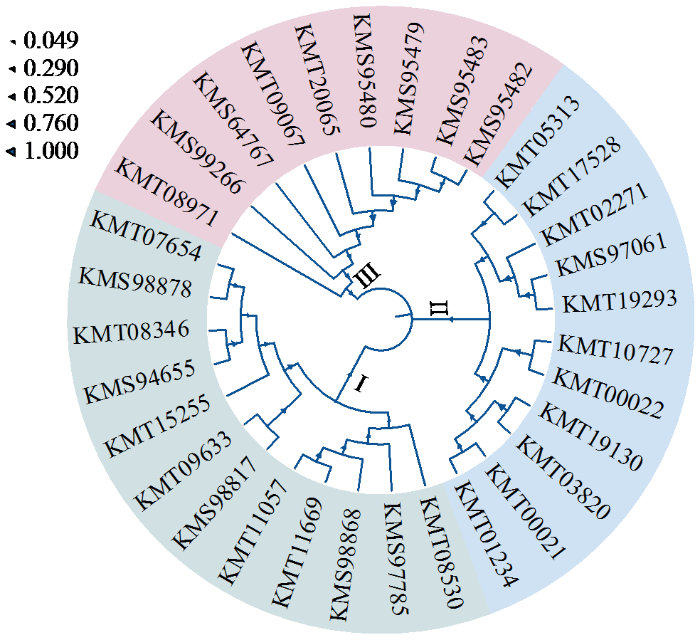
图1
甜菜C2H2型锌指蛋白家族进化树
Fig.1
Evolutionary tree of C2H2 zinc-finger protein family in sugar beet
各成员理论等电点介于5.00~11.72,包含11个酸性蛋白和21个碱性蛋白;蛋白氨基酸数量为88~1460;分子量大小在10 142.79~164 803.16 u;不稳定系数结果表明,除2个(BVRB_7g174490、BVRB_2g046350)不稳定系数小于40的C2H2型锌指蛋白外,其余均为不稳定蛋白。亚细胞定位预测发现,32个家族成员具有转录因子的特征,均定位于细胞核。其中,BVRB_9g212520同时被预测定位在细胞外,BVRB_042870在叶绿体和线粒体也有定位,BVRB_5g123490在高尔基体也有被预测到。32个家族成员均不含有信号肽序列,不属于分泌蛋白(表1)。
基因结构多样性和保守基序差异可能关系到多基因家族进化的机制[6],因此,对32个家族成员进行了基因结构和保守基序分析。基因结构分析结果(图2)显示,甜菜C2H2型锌指蛋白家族除KMS94655和KMT09067外,所有成员均存在上游和下游非编码区,KMS94655只有上游非编码区,KMT09067无上下游非编码区。整个家族中大部分成员只含有一个编码序列(coding sequence,CDS),部分成员CDS数量较多,其中KMT08530的数量最多,有12个CDS(图2d)。每个家族成员都至少含有1个锌指结构域,大多数成员具有zf-C2H2-6结构域,为典型的锌指结构域;第Ⅱ亚族的每个成员都含有2个及以上的zf-C2H2-6结构域。部分成员具有zf-C2H2、zf-C2H2_jaz和zf-C2H2_3rep,也都属于锌指结构域(图2c)。
图2

图2
甜菜C2H2型锌指蛋白家族成员的进化树(a)、保守基序(b)、结构域(c)和基因结构(d)分析
Fig.2
Phylogenetic tree (a), conserved motifs (b), domains (c) and gene structure (d) analysis of C2H2 zinc-finger protein family members in sugar beet
图3

图3
甜菜C2H2型锌指蛋白家族成员的保守基序序列
Fig.3
Conserved motif sequences of C2H2 zinc-finger protein family members in sugar beet
通过最大似然法构建了甜菜、拟南芥和水稻C2H2型锌指蛋白的系统发育树(图4)。系统发育分析结果表明,大多数甜菜的C2H2型锌指蛋白成员与拟南芥亲缘关系较近,KMT01234和AT4G35280.1、KMS98868和AT3G53820.1、KMT07654和AT1G10480.1互为亲缘关系较近的直系同源基因。
图4
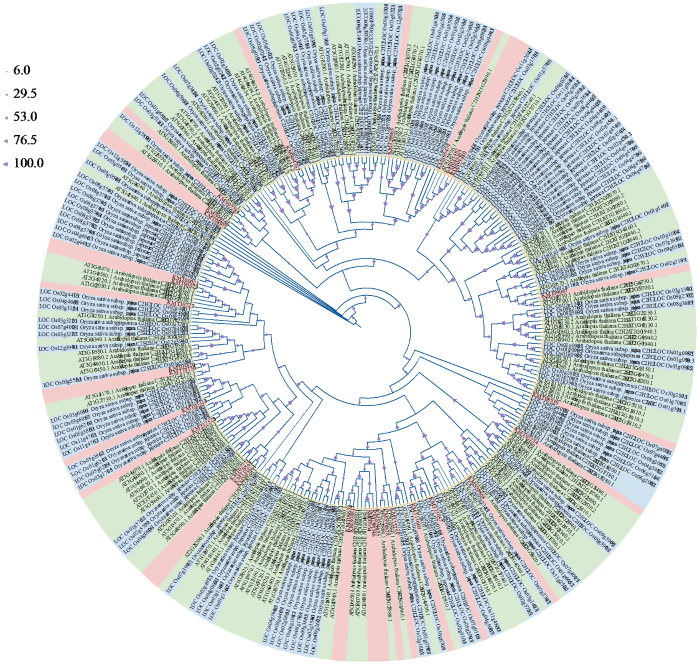
图4
甜菜、拟南芥、水稻的C2H2型锌指蛋白系统发育树
粉色为甜菜,绿色为拟南芥,蓝色为水稻。
Fig.4
C2H2 zinc-finger protein phylogenetic tree of sugar beet, Arabidopsis and rice
Pink is sugar beet, green is Arabidopsis, blue is rice.
2.2 甜菜C2H2型锌指蛋白家族成员的染色体定位
利用TBtools绘制甜菜C2H2锌指蛋白家族成员在染色体上的位置,部分C2H2转录因子定位在未完全拼接的染色体片段上(图5)。分析结果显示,部分染色体上存在多个转录本,其中6号染色体上定位到的转录本数量最多,有6个转录本。转录本位置与染色体基因密度没有明显相关性,大部分定位在基因密度较低的位置。
图5
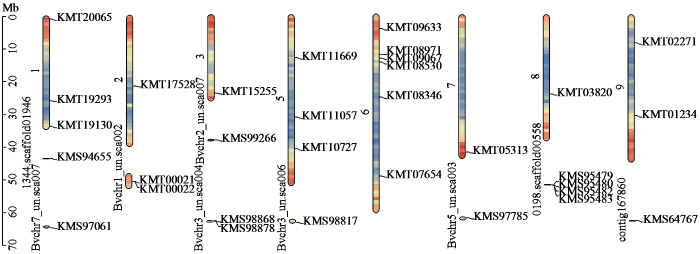
图5
甜菜C2H2型锌指蛋白家族成员的染色体定位
染色体颜色表示基因密度,颜色越红密度越高,颜色越蓝密度越低。
Fig.5
Chromosome location of C2H2 zinc-finger protein family members in sugar beet
Chromosome color indicates gene density, with higher density for redder colors and lower density for bluer colors.
2.3 启动子顺式作用元件分析
图6
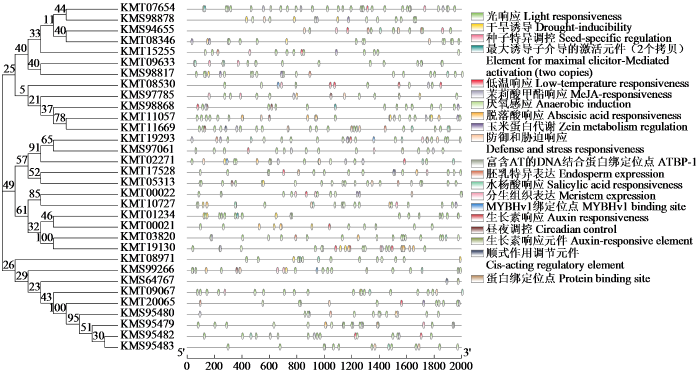
图6
甜菜C2H2型锌指蛋白转录因子启动子顺式作用元件分析
Fig.6
Promoter cis-element analysis of C2H2 zinc-finger protein transcription factor in sugar beet
2.4 镉胁迫下甜菜C2H2型锌指蛋白的表达
为探究不同浓度镉胁迫下甜菜C2H2锌指蛋白的表达情况,进行了0、1、3和5 mmol/L 4个CdCl2浓度处理下的转录组测序。结合转录组测序结果,共有29个家族成员在镉胁迫下差异表达,并根据其表达量绘制了热图(图7)。结果显示,大部分家族成员在未受到镉胁迫时表达水平较低,在受到镉胁迫后表达量呈上升趋势。但随胁迫浓度升高,部分家族成员的表达量有所下降,特别是在3 mmol/L浓度处理下,除BVRB_6g136840、BVRB_2g037310、BVRB_007850和BVRB_ 6g137660外所有家族成员表达量均下降。少部分家族成员表达量随胁迫处理浓度升高而逐渐升高,如BVRB_6g136840和BVRB_6g137660。
图7
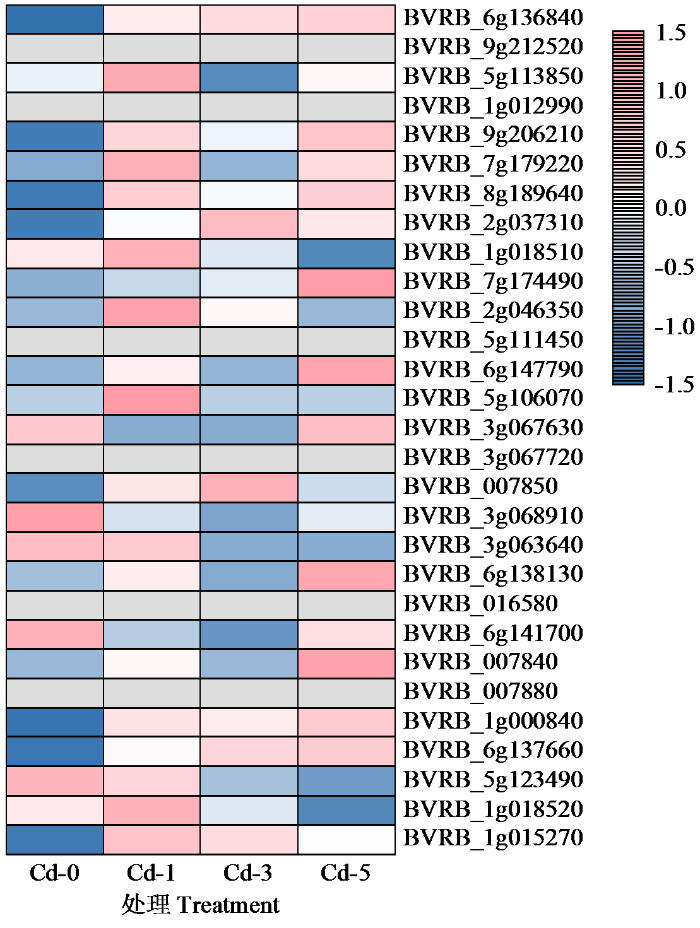
图7
甜菜C2H2型锌指转录因子表达热图
Fig.7
Heatmap of C2H2 zinc-finger transcription factor expression in sugar beet
2.5 甜菜C2H2锌指转录因子调控靶基因预测
为探究甜菜C2H2锌指转录因子与镉胁迫下差异表达基因的调控关系,利用PlantTFDB数据库对3组对比共有的差异基因上游2000 bp序列进行了转录因子结合位点的在线预测。根据预测结果筛选得到了4个甜菜C2H2型锌指转录因子家族成员(BVRB_6g137660、BVRB_8g189640、BVRB_ 1g000840、BVRB_007840),分别调控了45、44、32、18个差异表达基因(图8),且这4个成员在镉胁迫下表达量均不同程度提高。各成员调控的靶基因之间重叠部分较少,大部分靶基因仅受1个成员的调控。许多靶基因已被证实能够在盐胁迫、渗透胁迫、重金属胁迫等非生物胁迫下发挥作用,如胆碱单加氧酶(BVRB_6g146100)、铁蛋白(BVRB_ 5g103250)、金属耐受蛋白(BVRB_2g037080)、ABC转运蛋白(BVRB_5g109050)、富含亮氨酸的重复受体样丝氨酸/苏氨酸蛋白激酶(BVRB_6g153050)等。
图8
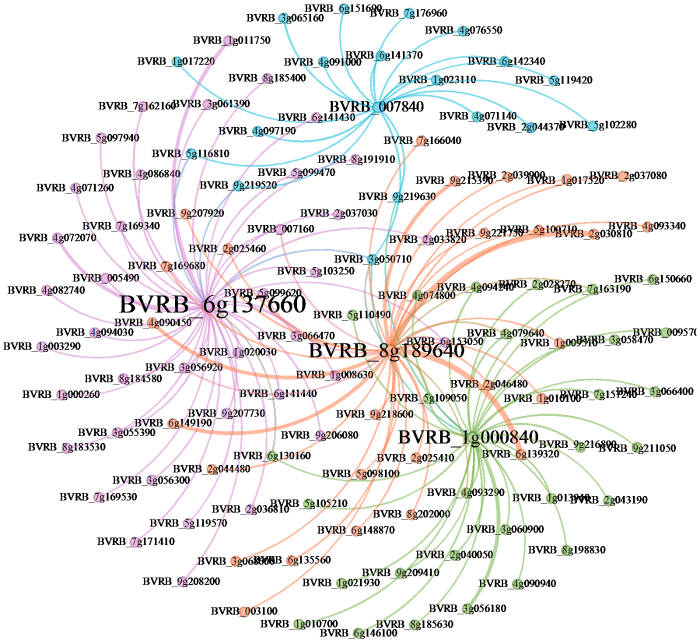
图8
甜菜C2H2型锌指转录因子与靶基因调控网络图
Fig.8
Regulatory network map of C2H2 zinc-finger transcription factors and target genes in sugar beet
3 讨论
本研究从甜菜基因组中鉴定到了32个C2H2型锌指蛋白转录因子,被分为3个亚族。理化性质分析结果表明,32个成员之间氨基酸数量和分子量大小差距较大,说明C2H2型锌指成员之间具有高度的复杂性。保守结构域及保守基序分析结果显示,motif 1具有很强的保守性,且motif 1和motif 2都含有的QALGGH基序是植物所特有的[20],该序列的保守性对锌指蛋白家族的DNA结合活性十分重要[21]。外显子和内含子的增加或减少可由基因片段的整合和重新排列引起,因此基因结构变异在基因家族的进化中起着重要作用[22]。基因结构分析结果显示,多数家族成员只有1个外显子,通常情况下植物的外显子和内含子数量都较少,因此才能根据环境变化迅速进行应答[9]。亚细胞定位预测结果显示,32个成员均在细胞核被定位到,符合转录因子在细胞核发挥作用的特点,而BVRB_9g212520、BVRB_042870、BVRB_5g123490这3个成员在其他位置也被预测到,说明他们与其他成员相比可能具有特殊的功能,但这仅是预测结果,各成员在甜菜内的具体定位仍需进一步验证。染色体定位结果显示,32个成员广泛分布于多个染色体和部分未完全拼接的染色体上,部分成员的位置排布较为紧密,说明这些成员之间可能相互关联,协同发挥作用[9]。C2H2型锌指蛋白不仅可以调控植物对各种胁迫做出响应,还参与了包括植物生长发育等一系列过程[6]。对32个家族成员启动子的顺式作用元件分析结果也体现了这一点,除与多种胁迫相关和受植物激素诱导的元件外,还存在部分调控植物生长发育的元件,这些元件能够调控下游靶基因的特异表达,从而应对各种环境变化。
为探究甜菜C2H2型锌指蛋白转录因子家族在镉胁迫下的表达模式,本研究进行了4个镉胁迫浓度下的转录组测序,表达分析热图显示,大部分家族成员在受到镉胁迫后表达量明显上升,说明这些C2H2型锌指蛋白转录因子在镉胁迫下被激活,可能继续调控下游靶基因的表达以应对镉胁迫。因此,本研究又进一步分析了C2H2型锌指蛋白转录因子与镉胁迫下差异表达基因间的调控关系。通过对3组处理共有差异基因的启动子序列进行转录因子结合位点的预测,我们发现部分共有差异表达基因的启动子序列上存在BVRB_6g137660、BVRB_ 8g189640、BVRB_1g000840、BVRB_007840这4个转录因子的结合位点。其中,BVRB_6g137660为锌指蛋白GAI相关因子1(zinc-finger protein GAI-ASSOCIATED FACTOR 1),又被称为不确定结构域蛋白2(protein indeterminate-domain 2),即IDD2。已有研究表明,IDD基因具有种子萌发[23]、开花调控[24]、淀粉代谢[25]、根系发育和氮代谢[26]等多种功能。BVRB_1g000840和BVRB_ 007840分别为质子根际毒性敏感蛋白1(STOP1)和质子根际毒性敏感蛋白2(STOP2),相关研究[27]表明,STOP1在土壤耐酸性机制和侧根生长方面发挥重要作用,并且在铝胁迫下,过表达该基因能够促进拟南芥根系生长。STOP2是STOP1的同源物,可以激活STOP1调控的一些基因的表达[28],但STOP2的表达水平远低于STOP1。BVRB_ 8g189640为锌指蛋白ZAT10(zinc-finger protein ZAT10),Dang等[15]发现,ZAT10在拟南芥对镉的吸收和解毒过程中起着双重作用,该基因参与了Cd摄取和Cd螯合及隔离的关键基因的调控,从而增强了植物对Cd胁迫的抗性,因此可将ZAT10列为提高植物耐镉性的潜在候选基因。
在受调控的靶基因中,部分基因已被证实能够在镉胁迫下发挥作用,例如BVRB_2g037080,为金属耐受蛋白(metal tolerance protein,MTP),在提高对重金属的耐受性和稳态方面起着重要作用[29]。有研究[30]表明,Cd胁迫下,在烟草和酵母中过表达MTP8可增强它们对Cd的耐受性,更好地应对由Cd胁迫引起的活性氧突增,降低对植物的伤害。BVRB_6g153050为富含亮氨酸的重复受体样丝氨酸/苏氨酸蛋白激酶,He等[31]在镉胁迫下对该基因进行了家族鉴定和分析,并将筛选出的枢纽基因Sa0F.522在酵母中异源表达,发现降低了其对镉胁迫的敏感性,证实了该基因在镉信号感知中的功能。BVRB_5g109050为ABC转运蛋白G家族成员(ABC transporter G family,ABCG),ABC转运体参与了金属离子的转运和解毒,OsPDR20(ABCG家族成员)积极参与了镉胁迫下水稻作物的镉积累和稳态[32]。综上所述,本研究初步探究了甜菜中C2H2型锌指蛋白转录因子的结构以及该基因家族在镉胁迫下与差异表达基因的应答关系,为深入探究C2H2型锌指蛋白转录因子以及甜菜种质改良提供了理论依据。
4 结论
通过对甜菜C2H2型锌指蛋白转录因子家族的全基因组鉴定,得到了32个家族成员,根据进化树和保守基序将整个家族划分为3个亚族,不同亚族间存在较大差异。通过镉胁迫下的表达分析筛选出了4个参与镉胁迫应答差异基因调控的C2H2型锌指转录因子。这4个转录因子在镉胁迫下表达量均不同程度上调,且调控金属耐受蛋白、ABC转运蛋白、富含亮氨酸的重复受体样丝氨酸/苏氨酸蛋白激酶等能够应答镉胁迫的基因,推测C2H2型锌指蛋白转录因子在甜菜对镉胁迫下的应答发挥重要作用。
参考文献
Genome-wide analysis of C2H2 zinc-finger gene family and its response to cold and drought stress in sorghum [Sorghum bicolor (L.) Moench]
C2H2 zinc-finger proteins:Master regulators of abiotic stress responses in plants
Conservation, diversification and expansion of C2H2 zinc-finger proteins in the Arabidopsis thaliana genome
DOI:10.1186/1471-2164-5-39
PMID:15236668
[本文引用: 1]

The classical C2H2 zinc finger domain is involved in a wide range of functions and can bind to DNA, RNA and proteins. The comparison of zinc finger proteins in several eukaryotes has shown that there is a lot of lineage specific diversification and expansion. Although the number of characterized plant proteins that carry the classical C2H2 zinc finger motifs is growing, a systematic classification and analysis of a plant genome zinc finger gene set is lacking.We found through in silico analysis 176 zinc finger proteins in Arabidopsis thaliana that hence constitute the most abundant family of putative transcriptional regulators in this plant. Only a minority of 33 A. thaliana zinc finger proteins are conserved in other eukaryotes. In contrast, the majority of these proteins (81%) are plant specific. They are derived from extensive duplication events and form expanded families. We assigned the proteins to different subgroups and families and focused specifically on the two largest and evolutionarily youngest families (A1 and C1) that are suggested to be primarily involved in transcriptional regulation. The newly defined family A1 (24 members) comprises proteins with tandemly arranged zinc finger domains. Family C1 (64 members), earlier described as the EPF-family in Petunia, comprises proteins with one isolated or two to five dispersed fingers and a mostly invariant QALGGH motif in the zinc finger helices. Based on the amino acid pattern in these helices we could describe five different signature sequences prevalent in C1 zinc finger domains. We also found a number of non-finger domains that are conserved in these families.Our analysis of the few evolutionarily conserved zinc finger proteins of A. thaliana suggests that most of them could be involved in ancient biological processes like RNA metabolism and chromatin-remodeling. In contrast, the majority of the unique A. thaliana zinc finger proteins are known or suggested to be involved in transcriptional regulation. They exhibit remarkable differences in the features of their zinc finger sequences and zinc finger arrangements compared to animal zinc finger proteins. The different zinc finger helix signatures we found in family C1 may have important implications for the sequence specific DNA recognition and allow inferences about the evolution of the members in this family.
Genome-wide identification of C2H 2 zinc-finger gene family in rice and their phylogeny and expression analysis
DOI:10.1007/s11103-007-9199-y
PMID:17610133
[本文引用: 1]
Transcription factors regulate gene expression in response to various external and internal cues by activating or suppressing downstream genes in a pathway. In this study, we provide a complete overview of the genes encoding C(2)H(2) zinc-finger transcription factors in rice, describing the gene structure, gene expression, genome localization, and phylogenetic relationship of each member. The genome of Oryza sativa codes for 189 C(2)H(2) zinc-finger transcription factors, which possess two main types of zinc-fingers (named C and Q). The Q-type zinc fingers contain a conserved motif, QALGGH, and are plant specific, whereas C type zinc fingers are found in other organisms as well. A genome-wide microarray based gene expression analysis involving 14 stages of vegetative and reproductive development along with 3 stress conditions has revealed that C(2)H(2) gene family in indica rice could be involved during all the stages of reproductive development from panicle initiation till seed maturation. A total of 39 genes are up-regulated more than 2-fold, in comparison to vegetative stages, during reproductive development of rice, out of which 18 are specific to panicle development and 12 genes are seed-specific. Twenty-six genes have been found to be up-regulated during three abiotic stresses and of these, 14 genes express specifically during the stress conditions analyzed while 12 are also up-regulated during reproductive development, suggesting that some components of the stress response pathways are also involved in reproduction.
The zinc-finger transcription factor SlZFP2 negatively regulates abscisic acid biosynthesis and fruit ripening in tomato
DOI:10.1104/pp.114.255174
PMID:25637453
[本文引用: 1]
Abscisic acid (ABA) regulates plant development and adaptation to environmental conditions. Although the ABA biosynthesis pathway in plants has been thoroughly elucidated, how ABA biosynthetic genes are regulated at the molecular level during plant development is less well understood. Here, we show that the tomato (Solanum lycopersicum) zinc finger transcription factor SlZFP2 is involved in the regulation of ABA biosynthesis during fruit development. Overexpression of SlZFP2 resulted in multiple phenotypic changes, including more branches, early flowering, delayed fruit ripening, lighter seeds, and faster seed germination, whereas down-regulation of its expression caused problematic fruit set, accelerated ripening, and inhibited seed germination. SlZFP2 represses ABA biosynthesis during fruit development through direct suppression of the ABA biosynthetic genes NOTABILIS, SITIENS, and FLACCA and the aldehyde oxidase SlAO1. We also show that SlZFP2 regulates fruit ripening through transcriptional suppression of the ripening regulator COLORLESS NON-RIPENING. Using bacterial one-hybrid screening and a selected amplification and binding assay, we identified the (A/T)(G/C)TT motif as the core binding sequence of SlZFP2. Furthermore, by RNA sequencing profiling, we found that 193 genes containing the SlZFP2-binding motifs in their promoters were differentially expressed in 2 d post anthesis fruits between the SlZFP2 RNA interference line and its nontransgenic sibling. We propose that SlZFP2 functions as a repressor to fine-tune ABA biosynthesis during fruit development and provides a potentially valuable tool for dissecting the role of ABA in fruit ripening. © 2015 American Society of Plant Biologists. All Rights Reserved.
Genome-wide analysis of C2H 2 zinc-finger family transcription factors and their responses to abiotic stresses in poplar (Populus trichocarpa)
The C2H2 zinc-finger protein SlZF3 regulates AsA synthesis and salt tolerance by interacting with CSN5B
DOI:10.1111/pbi.12863
PMID:29193661
[本文引用: 1]
Abiotic stresses are a major cause of crop loss. Ascorbic acid (AsA) promotes stress tolerance by scavenging reactive oxygen species (ROS), which accumulate when plants experience abiotic stress. Although the biosynthesis and metabolism of AsA are well established, the genes that regulate these pathways remain largely unexplored. Here, we report on a novel regulatory gene from tomato (Solanum lycopersicum) named SlZF3 that encodes a Cys2/His2-type zinc-finger protein with an EAR repression domain. The expression of SlZF3 was rapidly induced by NaCl treatments. The overexpression of SlZF3 significantly increased the levels of AsA in tomato and Arabidopsis. Consequently, the AsA-mediated ROS-scavenging capacity of the SlZF3-overexpressing plants was increased, which enhanced the salt tolerance of these plants. Protein-protein interaction assays demonstrated that SlZF3 directly binds CSN5B, a key component of the COP9 signalosome. This interaction inhibited the binding of CSN5B to VTC1, a GDP-mannose pyrophosphorylase that contributes to AsA biosynthesis. We found that the EAR domain promoted the stability of SlZF3 but was not required for the interaction between SlZF3 and CSN5B. Our findings indicate that SlZF3 simultaneously promotes the accumulation of AsA and enhances plant salt-stress tolerance.© 2017 The Authors. Plant Biotechnology Journal published by Society for Experimental Biology and The Association of Applied Biologists and John Wiley & Sons Ltd.
Molecular characterization and expression analysis reveal the roles of Cys2/His2 zinc-finger transcription factors during flower development of Brassica rapa subsp. chinensis
Genome-wide identification and analysis of the Q-type C2H2 gene family in potato (Solanum tuberosum L.)
Regulatory module WRKY33‐ ATL31‐IRT 1 mediates cadmium tolerance in Arabidopsis
Assessing the health risk of cadmium to the local population through consumption of contaminated vegetables grown in municipal solid waste- amended soil
DOI:10.1007/s10661-022-10104-w
PMID:35648302
[本文引用: 1]
Pollution caused by municipal solid waste (MSW) is becoming a serious threat to the environment. Composting may be an effective way to speed up the decomposition of biodegradable components in MSW, resulting in compost that can be utilized as an organic fertilizer. The pot experiments were carried out with different soil-MSW mixtures (100:0, 75:25, 50:50, and 25:75; w/w) to determine the impact of MSW on the bioconcentration of Cd in commonly consumed plants of Sargodha. The possible health risks were evaluated by applying pollution indices, such as the pollution load index, bioconcentration factor, enrichment factor, and health risk index. The pollution load index was higher than 1 in 75% MSW-amended soil. However, the concentration of Cd was found to be below the permissible limits in all studied vegetables, with a range of 0.019-0.106 mg/kg. In the study, serum samples from different volunteers living in four sites in Sargodha were also collected and analyzed. For vegetable crops, the health risk index (HRI) was less than one. It is concluded that the concentration of Cd was increased by increasing the fraction of MSW. Although the metal contents in the soil treated with MSW were not high enough to categorize the soil as polluted, these findings show that the reuse of MSW can serve as an alternative to mineral fertilizers. However, the presence of Cd in MSW can have a direct impact on soil fertility and, if biomagnified, on crop production and human health.© 2022. The Author(s), under exclusive licence to Springer Nature Switzerland AG.
Zinc-finger transcription factor ZAT 6 positively regulates cadmium tolerance through the glutathione-dependent pathway in Arabidopsis
ZAT10 plays dual roles in cadmium uptake and detoxification in Arabidopsis
The genome of the recently domesticated crop plant sugar beet (Beta vulgaris)
TBtools: An integrative toolkit developed for interactive analyses of big biological data
DOI:S1674-2052(20)30187-8
PMID:32585190
[本文引用: 1]
The rapid development of high-throughput sequencing techniques has led biology into the big-data era. Data analyses using various bioinformatics tools rely on programming and command-line environments, which are challenging and time-consuming for most wet-lab biologists. Here, we present TBtools (a Toolkit for Biologists integrating various biological data-handling tools), a stand-alone software with a user-friendly interface. The toolkit incorporates over 130 functions, which are designed to meet the increasing demand for big-data analyses, ranging from bulk sequence processing to interactive data visualization. A wide variety of graphs can be prepared in TBtools using a new plotting engine ("JIGplot") developed to maximize their interactive ability; this engine allows quick point-and-click modification of almost every graphic feature. TBtools is platform-independent software that can be run under all operating systems with Java Runtime Environment 1.6 or newer. It is freely available to non-commercial users at https://github.com/CJ-Chen/TBtools/releases.Copyright © 2020 The Author. Published by Elsevier Inc. All rights reserved.
MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets
DOI:10.1093/molbev/msw054
PMID:27004904
[本文引用: 1]
We present the latest version of the Molecular Evolutionary Genetics Analysis (Mega) software, which contains many sophisticated methods and tools for phylogenomics and phylomedicine. In this major upgrade, Mega has been optimized for use on 64-bit computing systems for analyzing larger datasets. Researchers can now explore and analyze tens of thousands of sequences in Mega The new version also provides an advanced wizard for building timetrees and includes a new functionality to automatically predict gene duplication events in gene family trees. The 64-bit Mega is made available in two interfaces: graphical and command line. The graphical user interface (GUI) is a native Microsoft Windows application that can also be used on Mac OS X. The command line Mega is available as native applications for Windows, Linux, and Mac OS X. They are intended for use in high-throughput and scripted analysis. Both versions are available from www.megasoftware.net free of charge.© The Author 2016. Published by Oxford University Press on behalf of the Society for Molecular Biology and Evolution. All rights reserved. For permissions, please e-mail: journals.permissions@oup.com.
AtZAT4,a C2H2-type zinc-finger transcription factor from Arabidopsis thaliana, is involved in pollen and seed development
C2H 2 zinc-finger proteins response to abiotic stress in plants
Divergence of duplicate genes in exon-intron structure
The naked endosperm genes encode duplicate indeterminate domain transcription factors required for maize endosperm cell patterning and differentiation
DOI:10.1104/pp.114.251413
PMID:25552497
[本文引用: 1]
The aleurone is the outermost layer of cereal endosperm and functions to digest storage products accumulated in starchy endosperm cells as well as to confer important dietary health benefits. Whereas normal maize (Zea mays [Zm]) has a single aleurone layer, naked endosperm (nkd) mutants produce multiple outer cell layers of partially differentiated cells that show sporadic expression of aleurone identity markers such as a viviparous1 promoter-β-glucuronidase transgene. The 15:1 F2 segregation ratio suggested that two recessive genes were involved, and map-based cloning identified two homologous genes in duplicated regions of the genome. The nkd1 and nkd2 genes encode the INDETERMINATE1 domain (IDD) containing transcription factors ZmIDDveg9 and ZmIDD9 on chromosomes 2 and 10, respectively. Independent mutant alleles of nkd1 and nkd2, as well as nkd2-RNA interference lines in which both nkd genes were knocked down, also showed the nkd mutant phenotype, confirming the gene identities. In wild-type kernels, the nkd transcripts were most abundant around 11 to 16 d after pollination. The NKD proteins have putative nuclear localization signals, and green fluorescent protein fusion proteins showed nuclear localization. The mutant phenotype and gene identities suggest that NKD controls a gene regulatory network involved in aleurone cell fate specification and cell differentiation. © 2015 American Society of Plant Biologists. All Rights Reserved.
Modulation of sugar metabolism by an INDETERMINATE DOMAIN transcription factor contributes to photoperiodic flowering in Arabidopsis
Inferring transcriptional gene regulation network of starch metabolism in Arabidopsis thaliana leaves using graphical Gaussian model
Making roots, shoots, and seeds: IDD gene family diversification in plants
DOI:S1360-1385(17)30204-2
PMID:29056440
[本文引用: 1]
The INDETERMINATE DOMAIN (IDD) family of transcriptional regulators controls a diversity of processes in a variety of plant tissues and organs and at different stages of plant development. Several recent reports describe the genetic characterization of IDD family members, including those that are likely to regulate C kranz anatomy, with implications for the engineering of C traits into C crops. In this review we summarize the reported functions of IDD members in the regulation of metabolic sensing and leaf, root, seed, and inflorescence development. We also provide an IDD phylogeny for the grasses and suggest future directions and strategies to define the function of IDDs in C photosynthesis and other developmental processes.Copyright © 2017 Elsevier Ltd. All rights reserved.
GhSTOP1,a C2H 2 type zinc- finger transcription factor is essential for aluminum and proton stress tolerance and lateral root initiation in cotton
DOI:10.1111/plb.12895
PMID:30098101
[本文引用: 1]
Aluminum (Al) and proton (H ) ions are major acid soil stress factors deleteriously affecting plant root growth and crop yield. In our preliminary studies, cotton (Gossypium hirsutum L.) seedlings displayed very sensitive phenotypes to Al and H rhizotoxicities. Based on previous Arabidopsis results, we aimed to characterise the Al stress responsive Sensitive to Proton rhizotoxicity 1 (GhSTOP1) transcription system in cotton using RNAi-mediated down-regulation. With the help of seed embryo apex explants, we developed transgenic cotton plants overexpressing a GhSTOP1-RNAi cassette with NPTII selection. Kanamycin-tolerant T1 seedlings were further considered for Al and H stress tolerance studies. Down-regulation of the GhSTOP1 increased sensitivity to Al and proton rhizotoxicities, and root growth was significantly reduced in RNAi lines. The expression profile of GhALMT1 (Aluminum-activated Malate Transporter 1), GhMATE (Multidrug and Toxic Compound Extrusion), GhALS3 (Aluminium Sensitive 3) and key genes involved in the GABA shunt were down-regulated in the transgenic RNAi lines. Additionally, the lateral root initiation process was delayed and expression of GhNAC1, which is involved in lateral roots, was also suppressed in transgenic lines. Besides, overexpression of GhSTOP1 in Arabidopsis accelerated root growth and AtMATE and AtALMT1 expression under Al stress conditions. These analyses indicate that GhSTOP1 is essential for the expression of several genes which are necessary for acid soil tolerance mechanisms and lateral root initiation.© 2018 German Society for Plant Sciences and The Royal Botanical Society of the Netherlands.
STOP2 activates transcription of several genes for Al-and low pH-tolerance that are regulated by STOP1 in Arabidopsis
DOI:10.1093/mp/sst116
PMID:23935008
[本文引用: 1]
The zinc-finger protein STOP1 (sensitive to proton rhizotoxicity 1) regulates transcription of multiple genes critical for tolerance to aluminum (Al) and low pH in Arabidopsis. We evaluated the contributions of genes that are suppressed in the stop1 mutant to Al- and low pH-tolerance using T-DNA-inserted disruptants, and transgenic stop1 mutants expressing each of the suppressed genes. STOP2, a STOP1 homolog, partially recovered Al- and low pH-tolerance by recovering the expression of genes regulated by STOP1. Growth and root tip viability under proton stress were partially rescued in the STOP2-complemented line. STOP2 localized in the nucleus and regulated transcription of two genes (PGIP1 and PGIP2) associated with cell wall stabilization at low pH. GUS assays revealed that STOP1 and STOP2 showed similar cellular expression in the root. However, the expression level of STOP2 was much lower than that of STOP1. In a STOP1 promoter::STOP2-complemented line, Al tolerance was slightly recovered, concomitant with the recovery of expression of ALS3 (aluminum sensitive 3) and AtMATE (Arabidopsis thaliana multidrug and toxic compound extrusion), while the expression of AtALMT1 (aluminum-activated malate transporter 1) was not recovered. These analyses indicated that STOP2 is a physiologically minor isoform of STOP1, but it can activate expression of some genes regulated by STOP1.
Comprehensive genome wide identification and expression analysis of MTP gene family in tomato (Solanum lycopersicum) under multiple heavy metal stress
DOI:10.1016/j.sjbs.2021.07.073
PMID:34866994
[本文引用: 1]
Plant metal tolerance proteins (MTPs) play major roles in enhancing resistance to heavy metal tolerance and homeostasis. However, the role of genes in tomato, which is one of the most popular crops, is still largely limited. Hence, we investigated genome-wide study of tomato, including phylogenetic, duplication, gene structure, gene ontology and previous transcriptomic data analysis. Moreover, the expression behaviour under various heavy metals stress has rarely been investigated. In the current study, eleven candidate genes were genome-wide identified and classified into three major groups; Mn-cation diffusion facilitators (CDFs), Fe/Zn-CDFs, and Zn-CDFs based on the phylogeny. Structural analysis of s showed high gene similarity within the same group with cation_efflux or ZT_dimerdomains. Evolutionary analysis revealed that segmental duplication contributed to the expansion of the family. Gene ontology further showed the vital roles of in metal-related processes. Tissue-specific expression profiling exhibited similar expression patterns in the same group, whereas gene expression varied among groups. The expression was evaluated after tomato treatments by five divalent heavy metals (Cd, Co, Mn, Zn and Fe). genes displayed differential responses in either plant leaves or roots under heavy metals treatments. Nine and ten responded to at least one metal ion treatment in leaves and roots, respectively. In addition,,,, and exhibited the highest expression responses in most of heavy metals treatments. Overall, our findings presented a standpoint on the evolution of and their evolution in tomato and paved the way for additional functional characterization under heavy metal toxicity.© 2021 Published by Elsevier B.V. on behalf of King Saud University.
Identification and comprehensive analysis of the characteristics and roles of leucine-rich repeat receptor-like protein kinase (LRR-RLK) genes in Sedum alfredii hance responding to cadmium stress
OsPDR20 is an ABCG metal transporter regulating cadmium accumulation in rice
DOI:10.1016/j.jes.2022.09.021
PMID:37923431
[本文引用: 1]
Cadmium (Cd) is a non-essential toxic heavy metal, seriously posing high environmental risks to human health. Digging genetic resources relevant to functional genes is important for understanding the metal absorption and accumulation in crops and bioremediation of Cd-polluted environments. This study investigated a functionally uncharacterized ATP binding cassette transporter G family (ABCG) gene encoding a Pleiotropic Drug Resistance 20 (PDR20) type metal transporter which is localized to the plasma membrane of rice. OsPDR20 was transcriptionally expressed in almost all tissues and organs in lifespan and was strongly induced in roots and shoots of young rice under Cd stress. Ectopic expression of OsPDR20 in a yeast mutant ycf1 sensitive to Cd conferred cellular tolerance with less Cd accumulation. Knockdown of OsPDR20 by RNA interference (RNAi) moderately attenuated root/shoot elongation and biomass, with reduced chlorophylls in rice grown under hydroponic medium with 2 and 10 µmol/L Cd, but led to more Cd accumulation. A field trial of rice grown in a realistic Cd-contaminated soil (0.40 mg/kg) showed that RNAi plants growth and development were also compromised compared to wild-type (WT), with smaller panicles and lower spikelet fertility but little effect on yield of grains. However, OsPDR20 suppression resulted in unexpectedly higher levels of Cd accumulation in rice straw including lower leaves and culm and grain. These results suggest that OsPDR20 is actively involved in Cd accumulation and homeostasis in rice crops. The increased Cd accumulation in the RNAi plants has the potential application in phytoremediation of Cd-polluted wetland soils.Copyright © 2022. Published by Elsevier B.V.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}