


高寒黑土区为我国的主要粮食生产区[12]。近年来粮食综合生产能力不断提高,但当前该地区作物种植以连作为主[13],集约化种植导致耕地出现一系列土壤健康问题。2016年,我国部分地区提出探索试行耕地轮作制度[14],这对于提升我国耕地质量、保障粮食安全、保护生态健康具有重要意义。有研究[15-16]表明,轮作可以提高粮食产量,同时促进生态系统健康发展。不同类型作物间存在相互关系,在一定程度上受环境因子影响,同时植物生长也受到多种因素制约。通过轮作可以在时间上增加作物多样性,并恢复地面与地下的正向互动[17],不仅可以最大化地利用水资源和养分,还可以最大化地利用生态位置和土壤的生物多样性[18]。轮作对于增强土壤生态系统的功能也具有巨大的潜力,并在维护农业系统中的土壤服务功能方面起到关键作用[19-20]。在中国南方地区,小麦与水稻、油菜以及玉米之间存在广泛而长期的轮作关系,这些轮作方式可能影响土壤微生物活性及其组分变化,进而影响土壤肥力状况。因此,更多元化的轮作策略提升了土壤微生物群落的代谢多样性和活性[5,17],同时也增加了作物的生物量和产量[21]。
本文以高寒黑土区大兴安岭西麓为研究区,该区域气候独特,属寒温带大陆性季风气候,有高寒禁区之称。因其气候湿润,夏季多降水,温差较大,年有效积温约2100 ℃,故受积温影响,大田种植主要作物有小麦、甜菜、油菜、水飞蓟、大麦与马铃薯,土壤则以黑钙土和草甸土为主,结构良好、质地疏松。其昼夜温差大,非常适宜马铃薯生产,地理位置也是国际上公认的马铃薯最佳种植带,因此将马铃薯作为当地优势作物,其余作物为其轮作倒茬作物。研究高寒黑土区不同轮作模式对土壤真菌群落结构的影响,对于加强黑土地保护、提高土壤结构稳定性具有重要意义,对实现黑土区耕地资源可持续利用和提高粮食产量至关重要。
1 材料与方法
1.1 试验地概况
试验地位于内蒙古自治区呼伦贝尔市海拉尔区哈克镇下辖社区谢尔塔拉农牧场(119°57′ E,49°25′ N),属寒温带大陆性季风气候,无霜期90~100 d,年均气温约-2.1 ℃,昼夜温差大,年均降水量约300 mm,降水集中在7-9月。
1.2 试验设计
采用大田试验,受无霜期限制,当地大面积种植的作物主要为小麦(Ta)、甜菜(Bv)、油菜(Bn)、水飞蓟(Sm)、大麦(Hv)与马铃薯(St),根据当地种植模式,设6种不同处理,包括2年和3年轮作处理,分别为T1(马铃薯―小麦―甜菜―马铃薯)、T2(马铃薯―水飞蓟―小麦―马铃薯)、T3(小麦―甜菜―小麦―马铃薯)、T4(油菜―小麦―甜菜―马铃薯),T5(小麦―油菜―大麦―马铃薯)和CK(油菜―小麦―马铃薯―马铃薯)。每个处理3次重复,于2022年5月初播种马铃薯,株距25 cm,行距90 cm,施肥用量、耕作措施、田间管理遵从当地种植习惯。
1.3 土壤样品采集
在马铃薯收获时(9月上旬),采用五点法随机选择植株,用抖落法采集0~20 cm耕层的根际土壤并均匀混合,将土样装入冰盒内的密封袋中,保存在实验室内-80 ℃冰箱中用于真菌群落的测定。
1.4 土壤DNA提取和高通量测序
将保存在-80 ℃冰箱中的土壤样品取出,对土壤样品基因组DNA进行提取。真菌ITS基因以引物ITS1-1F-F(5′-CTTGGTCATTTAGAGGAAGTA A-3′)和ITS1-1F-R(5′-GCTGCGTTCTTCATCGA TGC-3′)[22]进行PCR扩增。采用Illumina NovaSeq 6000平台进行测序分析。
1.5 数据处理
使用QIIME2 2021等软件对原始数据进行拼接和去嵌合体等步骤,以100%相似度聚类,通过DADA2平台质控后产生的每个去重的序列称为ASVs。基于ASV的丰度及注释信息,对每个样本在不同分类水平上序列的比例进行统计,使用Perl(V5.28.3)circos软件(V0.69-6)绘制不同分类水平物种圈图。土壤微生物群落丰度图使用R(V3.6.2)对组间进行t-检验分析,使用ggplot2程序包绘制差异物种箱线图。基于ASV相对丰度,使用软件LEfSe(
2 结果与分析
2.1 不同轮作模式下土壤真菌群落多样性分析
2.1.1 土壤真菌群落ITS DNA测序
表1 不同轮作模式土壤样本信息与真菌类别
Table 1
| 处理Treatment | 有效序列Effective sequences | 门Phylum | 纲Class | 目Order | 科Family | 属Genus | 种Species |
|---|---|---|---|---|---|---|---|
| T1 | 277 136 | 7 | 15 | 34 | 53 | 87 | 122 |
| T2 | 233 459 | 7 | 18 | 51 | 84 | 136 | 206 |
| T3 | 246 624 | 7 | 17 | 47 | 79 | 134 | 201 |
| T4 | 269 395 | 7 | 18 | 46 | 91 | 146 | 219 |
| T5 | 259 567 | 7 | 17 | 41 | 67 | 111 | 171 |
| CK | 246 624 | 6 | 16 | 40 | 67 | 120 | 192 |
| 合计Total | 1 572 105 | 7 | 22 | 72 | 146 | 300 | 598 |
图1
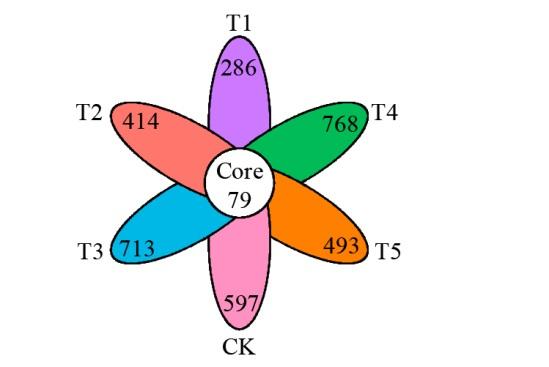
图1
不同轮作模式ASV分布花瓣图
Fig.1
Petal map of ASV distribution in different rotation patterns
ASV分布花瓣图可以用来统计样本之间共有和独有的物种数目,从而直观地显示样本在ASV水平上组成的相似性及重叠性。如图1所示,由花瓣图解析不同轮作模式和对照CK中真菌群落的异同,发现T1、T2、T3、T4、T5和CK处理的土壤真菌ASV数目分别286、414、713、768、493和597,共有ASV数目为79,占总数2.4%。综上,可通过高通量测序技术获得较多真菌群落种类和丰富度方面的信息。
2.1.2 土壤真菌群落α多样性分析
图2
α多样性指数可以反映微生物群落的丰度和多样性。如表2所示,与CK处理相比,T3、T4和T5处理土壤中真菌ACE指数、Chao1指数明显增加,表明3年轮作处理的真菌丰富度较高。T3和T4处理土壤中真菌的Simpson指数和Shannon指数较CK处理明显增加,表明2个处理中真菌多样性较高。T1和T2处理土壤中真菌ACE指数、Chao1指数、Simpson指数和Shannon指数较CK处理明显降低,表明2个处理的土壤真菌丰富度或均匀度明显下降。
表2 不同轮作模式根际土壤真菌群落α多样性分析
Table 2
| 处理Treatment | ACE | Chao 1 | Shannon | Simpson |
|---|---|---|---|---|
| T1 | 398.61 | 398.25 | 4.34 | 0.7758 |
| T2 | 394.43 | 394.06 | 4.86 | 0.8725 |
| T3 | 498.29 | 497.62 | 5.88 | 0.9351 |
| T4 | 585.18 | 585.39 | 6.28 | 0.9501 |
| T5 | 499.07 | 498.06 | 4.99 | 0.8640 |
| CK | 453.74 | 453.18 | 5.15 | 0.9068 |
2.1.3 土壤真菌群落β多样性分析
如图3所示,土壤真菌群落β多样性非度量多维尺度分析(NMDS)结果表明,轮作处理整体与CK处理距离较远,说明轮作与连作的土壤真菌群落结构存在明显差异。而T1、T4、T5与T2处理在第二维度有明显分离,表明不同作物轮作处理之间土壤真菌群落结构存在明显差异。
图3
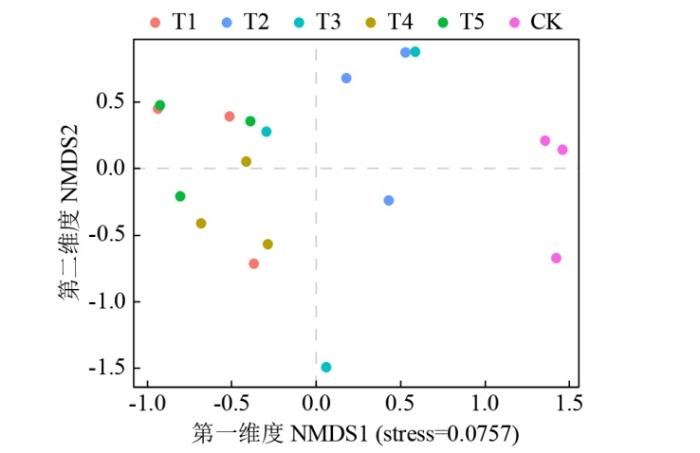
图3
不同轮作模式土壤真菌群落结构的非度量多维尺度分析
Fig.3
NMDS analysis of soil fungal community structure under different rotation patterns
2.2 不同轮作模式对土壤真菌群落结构特征的影响
2.2.1 土壤真菌群落结构组成
对不同轮作处理门水平前7位的真菌以及属水平前10位的真菌的相对丰度进行比较(图4)。对真菌而言,共检测到7门、22纲、72目、146科、300属和598种。其中,子囊菌门(Ascomycota)是真菌门水平的优势类群,相对丰度高达58.60%~82.66%。其次为担子菌门(Basidiomycota)(2.51%~12.35%)、接合菌门(Zygomycota)(2.63%~6.78%)、壶菌门(Chytridimycota)(0.03%~3.10%)、罗兹菌门(Rozellomycota)(0.01%~0.48%)、球孢菌门(Glomcromycota)(0.003%~0.09%)和芽枝菌门(Blastocladiomycota)(0.00%~0.05%)。未分类真菌(unclassified fungi)占真菌群落的4.49%~ 34.88%。其中,与CK处理相比,T1处理增加了子囊菌门;T1、T3和T4处理增加了接合菌门;T2、T3、T4和T5处理增加了球孢菌门;5种轮作处理均增加壶菌门和芽枝霉门,降低担子菌门的相对丰度(图4)。
图4
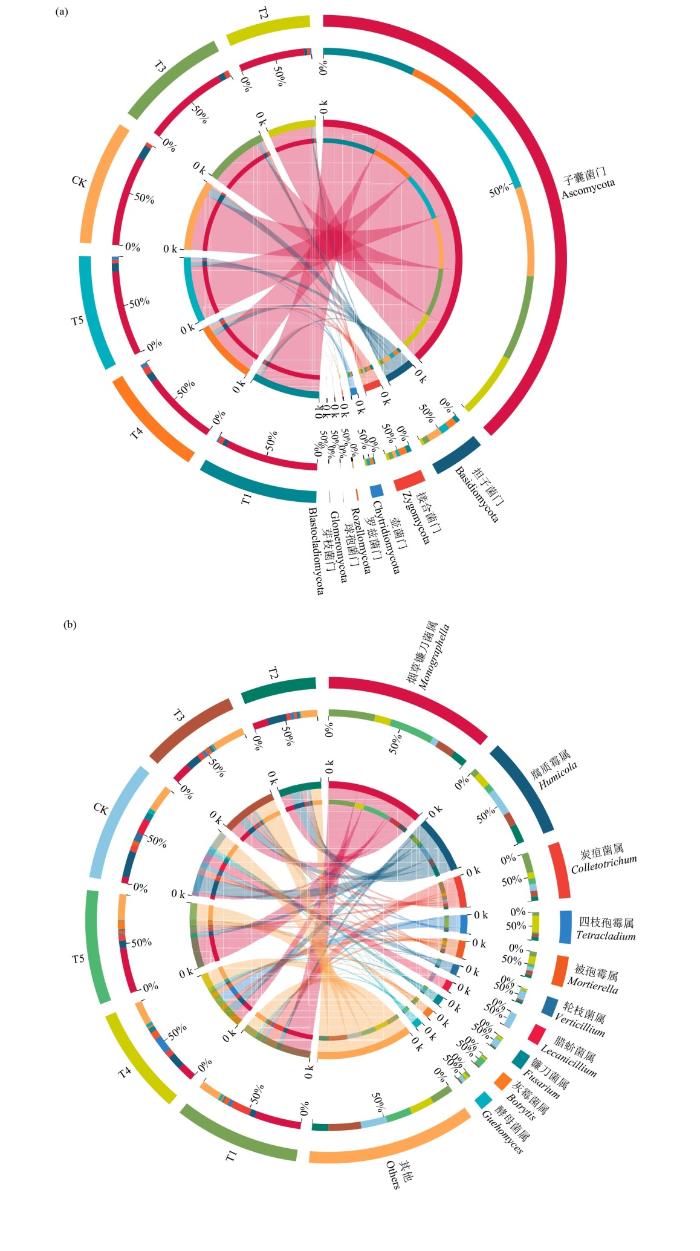
图4
不同轮作模式的门、属水平土壤真菌群落组成
Fig.4
Composition of soil fungal communities at the phylum and genus levels under different rotation patterns
在属水平上,不同轮作模式的土壤真菌群落均以烟草镰刀菌属(Monographella)、腐质霉属(Humicola)、炭疽菌属(Colletotrichum)和被孢霉属(Mortierella)为优势类群,相对丰度分别为4.98%~36.31%、4.71%~20.58%、3.42%~15.52%和2.15%~5.94%(图4)。此外,相对丰度大于1%的真菌属62个。与CK处理相比,5种轮作模式下烟草镰刀菌属的相对丰度增加,腐质霉属的相对丰度降低;炭疽菌属相对丰度仅T1处理增加;被孢霉属相对丰度仅T1、T3和T4处理增加。
2.2.2 土壤真菌群落结构差异
对不同轮作处理的土壤微生物群落进行PCoA分析(图5),发现PC1和PC2对土壤真菌群落结构的解释度分别为23.70%和15.06%,T1、T4和T5处理分布相对集中,与T2和CK处理分布相对分散,同时T2和CK处理的分布相对集中;Anosim分析表明,不同处理之间土壤真菌群落组成存在差异。
图5
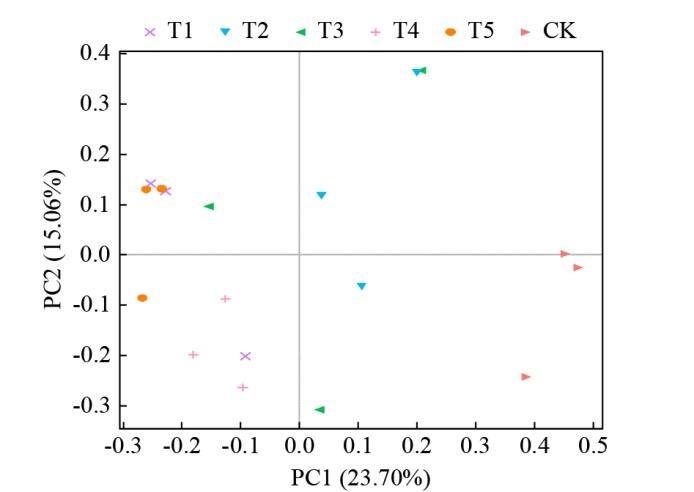
图5
不同轮作模式土壤真菌群落的PCoA分析
Fig.5
PCoA analysis of soil fungal communities under different rotation patterns
2.2.3 土壤真菌群落结构热图分析
如图6所示,门水平可分为2大类,其中T2处理为一支,子囊菌门在各处理中均为优势菌门;属水平分为2大类,其中T1和T5处理、T2和T4处理较为接近。
图6
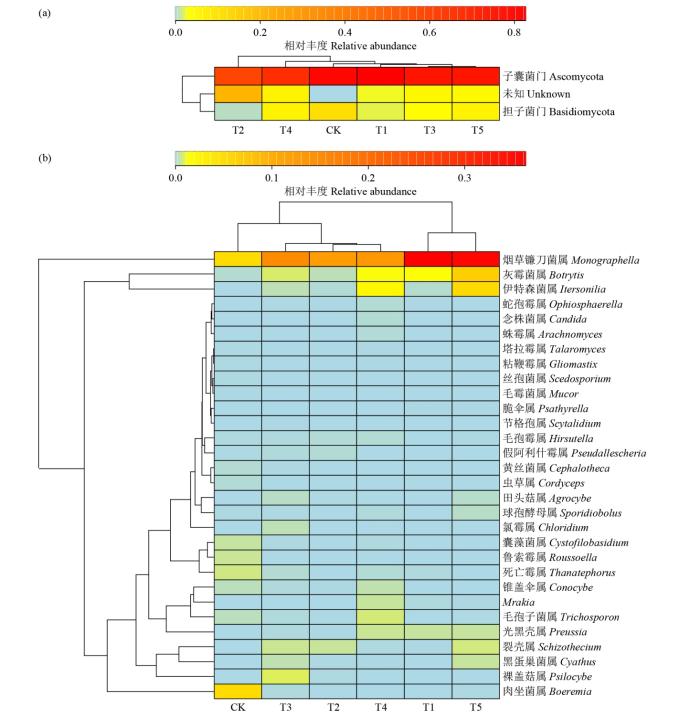
图6
不同轮作模式土壤真菌群落结构门、属水平的热图
Fig.6
Heat map of soil fungal community structure at the phylum and genus levels under different rotation patterns
2.2.4 土壤真菌群落组成差异
LEfSe(LDAEffect Size)代表了一种用于探索和阐释高维生物的标识,先检测找到具有显著丰度差异特征的类群,然后采用线性判别分析LDA来估算每个组分丰度对差异效果影响的大小。以LDA 4.0为阈值,对不同轮作模式土壤真菌群落组成进行线性判别分析,在真菌群落所有分类水平上发现了24个差异物种,T1处理包括6个生物标志物,T3处理包括3个生物标志物,T4处理包括3个生物标志物,T5处理包括7个生物标志物,而在T2处理中未发现差异物种(图7)。
图7
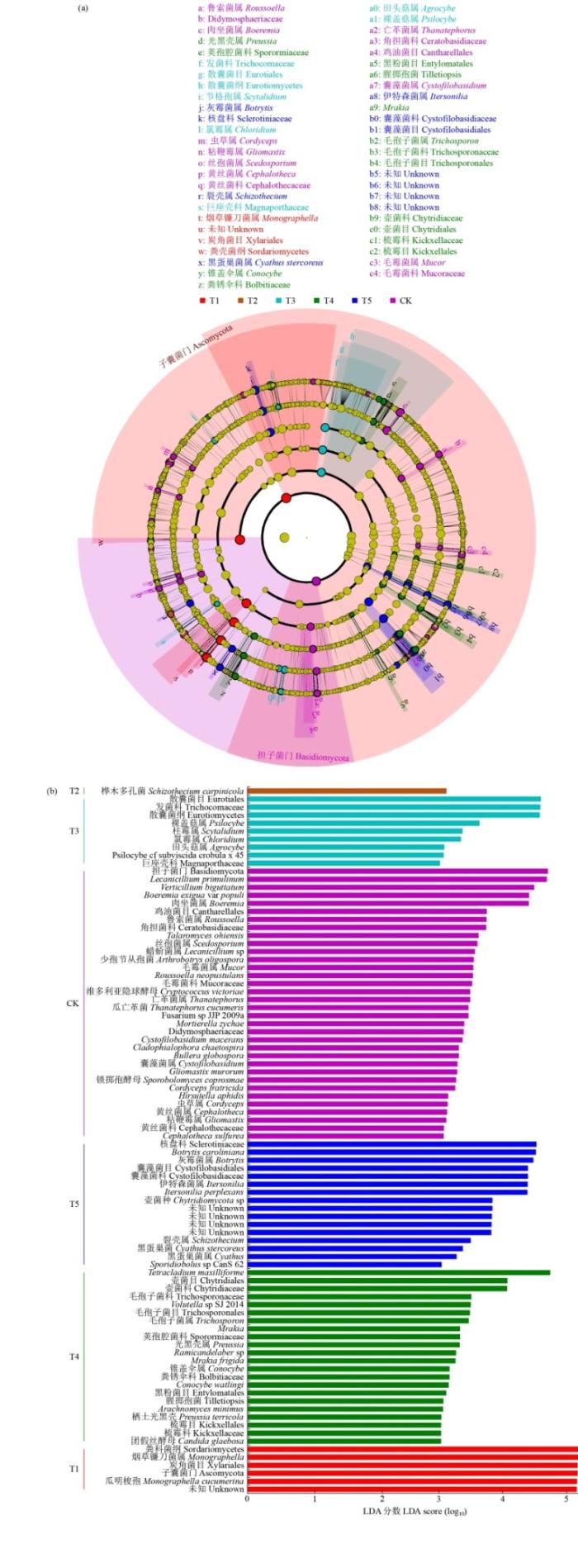
图7
土壤真菌群落的LEfSe差异分析和差异物种线性判别分析
Fig.7
LEfSe difference analysis and differential species linear discriminant analysis of soil fungal communities
2.3 不同轮作模式对土壤真菌群落的功能注释
根据FUNGuild功能预测对不同轮作模式真菌群落进行分类分析,从而了解和预测真菌群落功能。FUNGuild依据真菌的营养方式将真菌分为三大类:病理营养型(pathotroph)、共生营养型(symbiotroph)和腐生营养型(saprotroph)。
图8结果显示,不同轮作模式的真菌主要分为病理营养型、病理―腐生营养型、病理―腐生―共生营养型、病理―共生营养型、腐生营养型、腐生―共生营养型、共生营养型7个营养类型,其中病理营养型和腐生营养型占比较高,分别为26%~54%和19%~51%。与CK处理相比,T2、T3和T5处理增加了病理营养型、腐生营养型、病理―共生营养型和病理―腐生―共生营养型的相对丰度。
图8
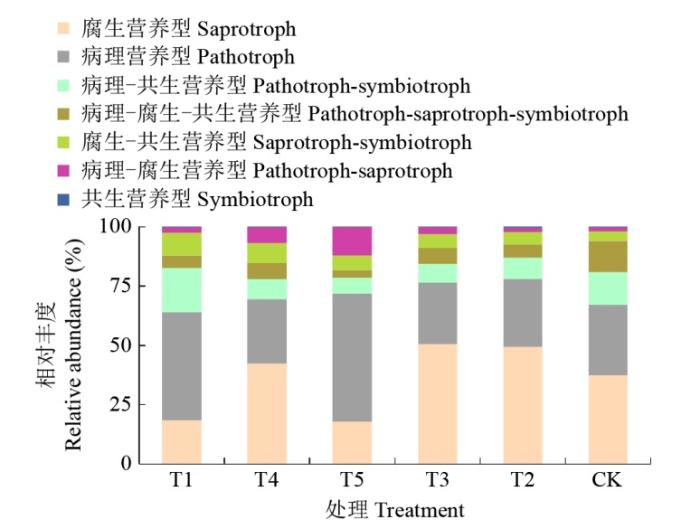
图8
不同轮作模式的土壤真菌营养方式相对丰度
Fig.8
Relative abundance of soil fungal nutrient patterns under different rotation patterns
3 讨论
3.1 不同轮作模式对马铃薯根际土壤真菌多样性的影响
微生物参与支持多个生态系统的功能运作[23],土壤中的微生物多样性被视为维护土壤健康和质量的核心因素[24],而土壤真菌多样性的增长可能对农业生态系统的功能运作和可持续性带来积极的影响[25]。真菌在土壤碳氮循环过程中扮演着关键角色,主要参与土壤中有机质的分解、腐殖质的形成、氨化过程以及团聚体的形成等[26]。因此,土壤真菌的多样性可以被视为评估土壤生态系统功能变化的一个关键指标[27-28]。有研究[5,10,29]表明,在轮作系统中增加作物种类导致土壤的物理化学特性和营养成分有所不同,从而影响土壤微生物群落,且轮作年限与微生物丰富度成正比;也有研究[30]表明,马铃薯连作较轮作处理真菌多样性变化不显著。本研究结果表明,T3和T4处理土壤真菌群落多样性和丰富度均有不同程度的升高,T5处理仅提高了土壤真菌ACE指数和Chao1指数,这表明轮作有助于提高土壤真菌α多样性,且T4处理的提升幅度最大,这一结果与牛倩云等[31]在谷子轮作土壤上的研究结果一致;但研究还发现T1和T2处理的土壤真菌群落多样性和丰富度均降低,T5处理的土壤真菌Shannon指数和Simpson指数均降低,这可能是由于作物根际的分泌物以及植物残体为微生物提供了充足的营养[32],而轮作中由于作物种类的增加,很大程度为土壤提供了更多更丰富的有机物来源[5,33],作物根系分泌物和含量具有独特性,不同生长阶段可能会产生不同类型和含量的根际分泌物,进而使根际土壤真菌多样性产生变化;另一方面可能是由于土壤水分、温度、养分水平和pH等土壤环境因素对根际分泌物的含量和组成的调控。因此,作物轮作对土壤真菌多样性的影响可能受多种根系分泌物和作物残留物共同作用。
3.2 不同轮作模式对马铃薯根际土壤真菌结构组成的影响
土壤微生物的群落组成在一定程度上能够反映土壤健康状况[34]。生态平衡的维护是多方面的,其中土壤真菌多样性及其群落结构起着重要的作用[35],通过土壤微环境的改变,影响真菌群落的组成和数量,进一步形成具有多样性和差异性的真菌群落特性[29,36-37]。不同轮作模式下真菌门水平主要优势菌群为子囊菌门。这一结果与前人的研究结果相似。子囊菌门是土壤中主要的分解者[38],作为真菌中最多的一个类群,包括腐生、寄生和共生3种[39],腐生真菌可以分解动植物残体,产生抗生素和有机酸,寄生真菌除引起植物病害,少数可寄生昆虫,在土壤生态系统中起重要作用[40]。子囊菌门能在抵抗环境压力,提高其在土壤中优势的同时,分解土壤中木质素和角质素等难降解的有机质,是养分循环和能量流动的主要驱动力[41],但由于受种植作物种类及环境的影响,其相对丰度也存在差异。Viaud等[42]研究发现农田中主要的真菌大部分是子囊菌门。本研究结果表明,在各处理中,子囊菌门为优势菌门,相对丰度最高,且在T1处理土壤中占绝对优势。5种轮作处理均增加了壶菌门和芽枝菌门的相对丰度,降低了担子菌门的相对丰度,这可能是因为马铃薯本身释放了与壶菌门、芽枝菌门有利的化合物和信号物质,不同作物的根系分泌物和残留物可能也提供了壶菌门和芽枝菌门所需的营养物质,进而促进它们的生长和繁殖,而壶菌门与担子菌门的竞争或抑制关系可能导致担子菌门的相对丰度降低。其他真菌门的规律不明显,这可能与种植作物的种类有关。
真菌各处理土壤群落前5位的属类,由大到小排序为烟草镰刀菌属、腐质霉属、炭疽菌属、四枝孢霉属和被孢霉属。T2、T3、T4和T5处理降低了炭疽菌属真菌的丰度,T1、T3和T5处理增加了被孢霉属真菌的丰度,其余轮作处理反之。有研究[43]指出,烟草镰刀菌属为常见的植物病原真菌属,是引起植物叶斑病的病原菌之一。腐质霉属可产生醋酸纤维素脱乙酰酶降解土壤中的生物质残体,增加土壤中木质素、纤维素和半纤维素含量[44]。炭疽菌属的寄主范围广,是一类全球分布的植物病原真菌[45]。被孢霉属大部分对提升土壤养分有效性以及改善土壤结构有利[46],而某些种属于病原性真菌,对植株幼苗生长有害[47],被孢霉属丰度过高可能会增加真菌性病害风险。四枝孢霉属在土壤中通常被归类为分解性真菌,具有较强的分解能力,可以有效降解和分解有机物质,如植物残体、根系和其他有机废弃物,进而提供更多的营养物质供植物吸收利用。同时,四枝孢霉属真菌与植物之间存在复杂的相互作用。一方面,四枝孢霉属真菌可以与植物根系建立共生关系,形成真菌根系共生,通过与植物根系的交换,促进植物的营养吸收和生长发育;另一方面,四枝孢霉属真菌也可能具有拮抗性,抑制土壤中一些病原真菌的生长,从而降低植物的病害风险。因此,这可能是轮作改变土壤质量的机制之一。
3.3 不同轮作模式对马铃薯根际土壤真菌群落功能的影响
真菌具有复杂的进化史,在土壤中存在多种营养方式,同时存在多种复合营养型[48]。本试验中子囊菌门是各处理真菌中种类最多的一个类群,包括腐生、寄生和共生,在土壤生态系统中起重要作用。本研究结果发现,T2、T3、T4处理以腐生营养型为主,其中T3处理中相对丰度最高,T1和T5处理则以病理营养型为主,而CK处理以腐生营养型为主,病理营养型为辅。其原因,一方面可能是因为腐生营养型真菌可以分解绝大部分有机质,如作物残体和动物粪便等,使其被吸收利用[48],如甜菜在深翻收获过程中将植物残渣和有机物质混入土壤更深层,并与土壤中的微生物接触,有助于加速有机质的分解,而小麦秸秆还田增加了土壤中有机质的含量,使腐生营养型真菌相对丰度增加;另一方面可能是因为不同轮作模式使土壤理化性状发生变化,导致土壤真菌营养类型发生改变。本研究中,T1和T5处理病理营养型的真菌相对丰度最高。这可能是由于小麦、油菜和大麦等作物释放的化合物,如根系分泌物和秸秆残体等,导致土壤真菌功能营养方式产生变化。
4 结论
采用高通量测序技术研究了高寒黑土区不同轮作模式下土壤真菌群落结构特征,结果显示,6种处理根际土壤样品中共检测到真菌7门、22纲、72目、146科、300属和598种。各处理土壤优势真菌门中,子囊菌门相对丰度最大,其次是担子菌门和接合菌门。与CK相比,T3、T4处理会提高土壤真菌多样性和丰富度,其中T4根际土壤真菌多样性和丰富度最高;不同轮作模式间土壤真菌群落营养类型的相对丰度不同,T2、T3和T4处理以腐生营养型为主,其中T3中相对丰度最高,T1和T5处理则以病理营养型为主。
参考文献
Belowground biodiversity and ecosystem functioning
Soil microbial diversity and agro-ecosystem functioning
Soil biodiversity and soil community composition determine ecosystem multifunctionality
Accounting for soil biotic effects on soil health and crop productivity in the design of crop rotations
DOI:10.1002/jsfa.6565
PMID:24408021
[本文引用: 1]

There is an urgent need for novel agronomic improvements capable of boosting crop yields while alleviating environmental impacts. One such approach is the use of optimized crop rotations. However, a set of measurements that can serve as guiding principles for the design of crop rotations is lacking. Crop rotations take advantage of niche complementarity, enabling the optimization of nutrient use and the reduction of pests and specialist pathogen loads. However, despite the recognized importance of plant-soil microbial interactions and feedbacks for crop yield and soil health, this is ignored in the selection and management of crops for rotation systems. We review the literature and propose criteria for the design of crop rotations focusing on the roles of soil biota and feedback on crop productivity and soil health. We consider that identifying specific key organisms or consortia capable of influencing plant productivity is more important as a predictor of soil health and crop productivity than assessing the overall soil microbial diversity per se. As such, we propose that setting up soil feedback studies and applying genetic sequencing tools towards the development of soil biotic community databases has a strong potential to enable the establishment of improved soil health indicators for optimized crop rotations.© 2014 Society of Chemical Industry.
The impact of crop rotation on soil microbial diversity: a meta-analysis
Diversifying crop rotation increased metabolic soil diversity and activity of the microbial community
The role of soil microorganisms in plant mineral nutrition—current knowledge and future directions
Rhizosphere microbiome structure alters to enable wilt resistance in tomato
Global diversity and geography of soil fungi
基于地学信息图谱的东北黑土区种植模式分析
DOI:10.13287/j.1001-9332.202203.017
[本文引用: 1]
不同种植模式具有不同的经济学和生态学意义,因地制宜确立合理的种植模式是保护黑土地的重要措施。目前,对东北黑土区的种植模式类型及其空间格局尚缺乏系统的认识。本研究基于东北黑土区2017—2019年作物分类数据,应用地学信息图谱理论和方法构建种植模式图谱,以揭示黑土区种植模式类型及其空间格局,并在此基础上使用分布指数模型和双变量空间自相关方法分析了种植模式的分布规律。结果表明: 1) 东北黑土区种植模式主要有玉米连作、水稻连作、大豆连作和米(玉米)豆(大豆)轮作,面积占比分别为38.3%、18.5%、10.3%、26.0%。其中,米豆轮作又包括米豆两年轮作、米米豆三年轮作、豆豆米三年轮作,分别占轮作面积的44.1%、34.5%、21.4%;2) 各类种植模式的水平地带性分异规律显著。其中,玉米连作在温度和湿度上的梯度分异特征均非常明显,水稻连作仅在湿度上的梯度分异特征非常明显,大豆连作和米豆轮作在温度和湿度上的梯度分异特征均不明显;3) 各类种植模式的空间集聚特征显著,种植模式间存在“大豆连作→豆豆米三年轮作→米豆两年轮作→米米豆三年轮作→玉米连作”的空间过渡特征。东北黑土区种植模式以连作为主,其分布规律可为今后轮作空间优化提供依据。
Increasing crop heterogeneity enhances multitrophic diversity across agricultural regions
National food production stabilized by crop diversity
Crop rotational diversity enhances belowground communities and functions in an agroecosystem
DOI:10.1111/ele.12453
PMID:26011743
[本文引用: 2]
Biodiversity loss, an important consequence of agricultural intensification, can lead to reductions in agroecosystem functions and services. Increasing crop diversity through rotation may alleviate these negative consequences by restoring positive aboveground-belowground interactions. Positive impacts of aboveground biodiversity on belowground communities and processes have primarily been observed in natural systems. Here, we test for the effects of increased diversity in an agroecosystem, where plant diversity is increased over time through crop rotation. As crop diversity increased from one to five species, distinct soil microbial communities were related to increases in soil aggregation, organic carbon, total nitrogen, microbial activity and decreases in the carbon-to-nitrogen acquiring enzyme activity ratio. This study indicates positive biodiversity-function relationships in agroecosystems, driven by interactions between rotational and microbial diversity. By increasing the quantity, quality and chemical diversity of residues, high diversity rotations can sustain soil biological communities, with positive effects on soil organic matter and soil fertility. © 2015 John Wiley & Sons Ltd/CNRS.
Anthropogenic drivers of soil microbial communities and impacts on soil biological functions in agroecosystems
Crop rotation complexity regulates the decomposition of high and low quality residues
Increasing cropping system diversity balances productivity, profitability and environmental health
Rhizosphere microbial communities explain positive effects of diverse crop rotations on maize and soybean performance
Detection of fungal DNA in human body fluids and tissues during a multistate outbreak of fungal meningitis and other infections
DOI:10.1128/EC.00046-13
PMID:23457192
[本文引用: 1]
Exserohilum rostratum was the major cause of an outbreak of fungal infections linked to injections of contaminated methylprednisolone acetate. Because almost 14,000 persons were exposed to product that was possibly contaminated with multiple fungal pathogens, there was unprecedented need for a rapid throughput diagnostic test that could detect both E. rostratum and other unusual agents of fungal infection. Here we report development of a novel PCR test that allowed for rapid and specific detection of fungal DNA in cerebrospinal fluid (CSF), other body fluids and tissues of infected individuals. The test relied on direct purification of free-circulating fungal DNA from fluids and subsequent PCR amplification and sequencing. Using this method, we detected Exserohilum rostratum DNA in 123 samples from 114 case-patients (28% of 413 case-patients for whom 627 samples were available), and Cladosporium DNA in one sample from one case-patient. PCR with novel Exserohilum-specific ITS-2 region primers detected 25 case-patients with samples that were negative using broad-range ITS primers. Compared to fungal culture, this molecular test was more sensitive: of 139 case-patients with an identical specimen tested by culture and PCR, E. rostratum was recovered in culture from 19 (14%), but detected by PCR in 41 (29%), showing a diagnostic sensitivity of 29% for PCR compared to 14% for culture in this patient group. The ability to rapidly confirm the etiologic role of E. rostratum in these infections provided an important contribution in the public health response to this outbreak.
An Underground Revolution: Biodiversity and Soil Ecological Engineering for Agricultural Sustainability
Metagenomic profiling of rhizosphere microbial community structure and diversity associated with maize plant as affected by cropping systems
Soil biodiversity for agricultural sustainability
Plant species and soil type cooperatively shape the structure and function of microbial communities in the rhizosphere
DOI:10.1111/j.1574-6941.2009.00654.x
PMID:19243436
[本文引用: 1]
The rhizosphere is of central importance not only for plant nutrition, health and quality but also for microorganism-driven carbon sequestration, ecosystem functioning and nutrient cycling in terrestrial ecosystems. A multitude of biotic and abiotic factors are assumed to influence the structural and functional diversity of microbial communities in the rhizosphere. In this review, recent studies on the influence of the two factors, plant species and soil type, on rhizosphere-associated microbial communities are discussed. Root exudates and the response of microorganisms to the latter as well as to root morphology were shown to shape rhizosphere microbial communities. All studies revealed that soil is the main reservoir for rhizosphere microorganisms. Many secrets of microbial life in the rhizosphere were recently uncovered due to the enormous progress in molecular and microscopic tools. Physiological and molecular data on the factors that drive selection processes in the rhizosphere are presented here. Furthermore, implications for agriculture, nature conservation and biotechnology will also be discussed.
How soil biota regulate C cycling and soil C pools in diversified crop rotations
Wheat,maize and sunflower cropping systems selectively influence bacteria community structure and diversity in their and succeeding crop's rhizosphere
Diversity and co-occurrence patterns of soil bacterial and fungal communities in seven intercropping systems
DOI:10.3389/fmicb.2018.01521
PMID:30034385
[本文引用: 1]
Intercropping plays a vital role in greenhouse production, and affects soil physicochemical properties and soil microbial communities structure, but influences of intercropping on the relationship of microorganisms are reported in continuous cropping soil rarely. Here, we investigated the effects of seven intercropping systems [alfalfa (Medicago sativa L.)/cucumber, trifolium (Trifolium repens L.)/cucumber, wheat (Triticum aestivum L.)/cucumber, rye (Secale cereale L.)/cucumber, chrysanthemum (Chrysanthemum coronrium L.)/cucumber, rape (Brassica campestris L.)/cucumber, mustard (Brassica juncea L.)/cucumber] on soil bacterial and fungal communities compared to the cucumber continuous cropping system in the greenhouse. The results showed that intercropping increased microbial OTU richness and fungal communities diversity, soil bacterial communities diversity was abundant in the trifolium-cucumber and mustard-cucumber systems. Nevertheless, there was no significant differences of microbial communities structure between intercropping and monoculture systems. Redundancy analysis indicated that soil microbial communities composition was indirectly influenced by soil properties. In addition, network analysis demonstrated that simple inter-relationships of fungal taxa were observed in the intercropping soil, and trifolium, wheat, and mustard intercropping systems had a complex connection between bacterial taxa. Taken together, trifolium and mustard as the intercrops significantly increased cucumber continuous cropping soil bacterial and fungal communities diversity. Moreover, intercropping strongly changed the relationships of microbial taxa, though did not shape notably soil microbial communities structure.
The Ascomycota tree of life: a phylum-wide phylogeny clarifies the origin and evolution of fundamental reproductive and ecological traits
DOI:10.1093/sysbio/syp020
PMID:20525580
[本文引用: 1]
We present a 6-gene, 420-species maximum-likelihood phylogeny of Ascomycota, the largest phylum of Fungi. This analysis is the most taxonomically complete to date with species sampled from all 15 currently circumscribed classes. A number of superclass-level nodes that have previously evaded resolution and were unnamed in classifications of the Fungi are resolved for the first time. Based on the 6-gene phylogeny we conducted a phylogenetic informativeness analysis of all 6 genes and a series of ancestral character state reconstructions that focused on morphology of sporocarps, ascus dehiscence, and evolution of nutritional modes and ecologies. A gene-by-gene assessment of phylogenetic informativeness yielded higher levels of informativeness for protein genes (RPB1, RPB2, and TEF1) as compared with the ribosomal genes, which have been the standard bearer in fungal systematics. Our reconstruction of sporocarp characters is consistent with 2 origins for multicellular sexual reproductive structures in Ascomycota, once in the common ancestor of Pezizomycotina and once in the common ancestor of Neolectomycetes. This first report of dual origins of ascomycete sporocarps highlights the complicated nature of assessing homology of morphological traits across Fungi. Furthermore, ancestral reconstruction supports an open sporocarp with an exposed hymenium (apothecium) as the primitive morphology for Pezizomycotina with multiple derivations of the partially (perithecia) or completely enclosed (cleistothecia) sporocarps. Ascus dehiscence is most informative at the class level within Pezizomycotina with most superclass nodes reconstructed equivocally. Character-state reconstructions support a terrestrial, saprobic ecology as ancestral. In contrast to previous studies, these analyses support multiple origins of lichenization events with the loss of lichenization as less frequent and limited to terminal, closely related species.
Estimating the phanerozoic history of the Ascomycota lineages: combining fossil and molecular data
DOI:10.1016/j.ympev.2014.04.024
PMID:24792086
[本文引用: 1]
The phylum Ascomycota is by far the largest group in the fungal kingdom. Ecologically important mutualistic associations such as mycorrhizae and lichens have evolved in this group, which are regarded as key innovations that supported the evolution of land plants. Only a few attempts have been made to date the origin of Ascomycota lineages by using molecular clock methods, which is primarily due to the lack of satisfactory fossil calibration data. For this reason we have evaluated all of the oldest available ascomycete fossils from amber (Albian to Miocene) and chert (Devonian and Maastrichtian). The fossils represent five major ascomycete classes (Coniocybomycetes, Dothideomycetes, Eurotiomycetes, Laboulbeniomycetes, and Lecanoromycetes). We have assembled a multi-gene data set (18SrDNA, 28SrDNA, RPB1 and RPB2) from a total of 145 taxa representing most groups of the Ascomycota and utilized fossil calibration points solely from within the ascomycetes to estimate divergence times of Ascomycota lineages with a Bayesian approach. Our results suggest an initial diversification of the Pezizomycotina in the Ordovician, followed by repeated splits of lineages throughout the Phanerozoic, and indicate that this continuous diversification was unaffected by mass extinctions. We suggest that the ecological diversity within each lineage ensured that at least some taxa of each group were able to survive global crises and rapidly recovered. Copyright © 2014 The Authors. Published by Elsevier Inc. All rights reserved.
Diversity of soil fungi studied by PCR-RFLP of ITS
一株特异腐质霉纤维素酶高产突变株的鉴定分析
DOI:10.19586/j.2095-2341.2019.0012
[本文引用: 1]
高温真菌特异腐质霉(Humicola insolens)具有强大的纤维素酶分泌和纤维素降解能力,因此在生物质能源、饲料和纺织工业等领域中具有良好的应用前景。在特异腐质霉T-DNA随机插入突变体库中筛选得到一株纤维素酶高产菌株T53。在纤维素诱导培养条件下,突变株T53发酵上清中的滤纸酶活力、内切β-1,4葡聚糖酶活力、纤维二糖水解酶活力、β-葡萄糖苷酶活力以及木聚糖酶活力较野生型菌株分别提高了40%、60%、90%、10%、60%。蛋白电泳结果显示,T53菌株的胞外总蛋白特别是纤维素酶与半纤维素酶的分泌量较野生型菌株有显著提高。TAIL-PCR克隆鉴定了T53菌株中的T-DNA插入位点侧翼序列,结果表明T-DNA插入位点位于一个可能的C2H2型锌指蛋白类转录因子编码基因的上游启动子区域。这为揭示突变株T53中纤维素酶的高产分子机制奠定了重要基础,有助于理解特异腐质霉中纤维素酶的表达调控机制,为特异腐质霉纤维素酶高产工程菌的构建奠定了理论和技术基础。
Interactions between arbuscular mycorrhizal fungi and phosphate-solubilizing fungus (Mortierella sp.) and their effects on Kostelelzkya virginica growth and enzyme activities of rhizosphere and bulk soils at different salinities
Dieback of Pinus nigra seedlings caused by a strain of

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}